Download presentation
Presentation is loading. Please wait.

1
Overview of Good Clinical Practice
care Overview of Good Clinical Practice community Donna W. Dorozinsky, RN, MSN, CCRC research teach

2
Welcome Please silence cell phones Please limit exits and entrances

3
Course Objectives Discuss the purpose and various sponsors of clinical research Discuss the evolution of Good Clinical Practice and the importance of it in today’s research environment. Discuss the general principles of Good Clinical Practice including ICH Guidelines, Title 45 and 21 CFR, Parts 11, 50, 54, 56, 312 and 314 Identify the sponsor, investigator and monitor responsibilities in conduct of FDA regulated studies.

4
The Clinical Research Process
Clinical trials are a principled partnership between the clinical investigator and industry Provide an avenue for providing cutting edge health care to your patients. Provide a scientific and ethical pathway for new product development. Create public awareness of the clinical research process.

5
The Clinical Research Process
Industry, in collaboration with clinical investigators, develops diagnostic, therapeutic, and preventative products to better serve the health care community. The clinical investigator plays an important role by conducting Clinical trials to further profile the new product by testing the safety and efficacy in a diversified patient population Safety and efficacy testing involving patients Conducting translational research

6
Clinical trial: What does it mean?
Trials to evaluate the effectiveness and safety of medications or medical devices by monitoring their effects on large groups of people.

7
Who are the players? Sponsor NIH CRO SMO Clinical Investigator
Institutional Review Board

8
The Sponsor Individual, group or company that takes responsibility for the design, management and financing of the study. Investigator-sponsored research Commercial organization Biotech company Pharmaceutical company Federally funded (NIH) The responsibilities of the sponsor can be retained by the original party or transferred to another organization such as a Clinical Research Organization (CRO).
 The responsibilities of the sponsor can be retained by the original party or transferred to another organization such as a Clinical Research Organization (CRO).")
9
National Institutes of Health
Department of Health and Human Services Funds research, conducts studies, and funds multicenter national studies Composed of 27 Institutes and Centers NIH annually invests over $28 billion in medical research through competitive grants 10% research is conducted at NIH in Bethesda, Maryland Examples National Cancer Institute

10
NIH Impacting Health Care
Death rates from heart disease and stroke fell by 40% and 51%, respectively, between 1975 and 2000. The overall five-year survival rate for childhood cancers rose to nearly 80% during the 1990s from under 60% in the 1970s. The number of AIDS-related deaths fell by about 70% between 1995 and 2001. Sudden infant death syndrome rates fell by more than 50% between 1994 and 2000. Infectious diseases—such as rubella, whooping cough, and pneumococcal pneumonia—that once killed and disabled millions of people are now prevented by vaccines. Quality of life for 19 million Americans suffering with depression has improved as a result of more effective medication and psychotherapy.

11
NIH Medical Discoveries
The sequencing of the human genome Bioterrorism research Aggressively pursue ways to make effective vaccines for deadly diseases like HIV/AIDS, tuberculosis, malaria, and potential agents of bioterrorism. Progress in understanding the immune system may lead to new ways to treat and cure diabetes, arthritis, asthma and allergies. New, more precise ways to treat cancer are emerging, such as drugs that zero in on abnormal proteins in cancer cells. Novel research methods are being developed that can identify the causes of outbreaks, such as Severe Acute Respiratory Syndrome (SARS), in weeks rather than months or years.
, in weeks rather than months or years.")
12
CRO Sponsor delegates specific responsibilities to the CRO
Accountability remains with the original sponsor. A CRO that assumes a sponsor obligation is required to comply with the local regulatory requirements Examples

13
Site Management Organization (SMO)
Individual, a network of individuals or an organization that sub-contracts clinical trial responsibilities from a CRO. Contract negotiations with the trial institution IRB approval Patient recruitment Patient follow-up Informed consent form (ICF) translation into vernacular languages Site initiation and trial close-out operations Trial-related documents archival and maintenance Reporting Serious Adverse Events to the CRO and the IRB/IEC
 translation into vernacular languages. Site initiation and trial close-out operations. Trial-related documents archival and maintenance. Reporting Serious Adverse Events to the CRO and the IRB/IEC.")
14
Clinical Investigator
Ensures that an investigation is conducted according to: the signed investigator statement, the investigational protocol applicable regulations; Protects the rights, safety, and welfare of subjects under the investigator's care Responsible for the control of drugs under investigation.

15
IRB A group of scientists, doctors, clergy, and consumers
Designed to protect study participants. Review and must approve the consent, protocol and any other information that is given or seen by the subject. Confirm that the trial is well designed, does not involve undue risks, and includes safeguards for subjects

16
What regulates this entire process?
The Federal Food, Drug and Cosmetic Act (505(a)) “…evidence consisting of adequate and well-controlled clinical investigations, by experts (education, training, and experience) to show the drug has the effect it claims.” 21 CFR
) …evidence consisting of adequate and well-controlled clinical investigations, by experts (education, training, and experience) to show the drug has the effect it claims. 21 CFR")
17
Good Clinical Practice
What is it? A collection of regulations, guidelines, and accepted quality research practices which define the standards and procedures for the design, conduct, performance, monitoring, auditing, recording, analysis, and reporting of clinical trials.

18
GCP Applicability All phases of clinical research
All aspects of clinical research Ethical conduct Study conduct SOPs Processing of data Archiving of records Quality assurance Staff training

19
History of GCP 1902 Biologics Act 1906 Food and Drug Act
1937 Elixir of Sulfanilamide Tragedy 1938 Food, Drug, and Cosmetics Act 1962 Thalidomide Tragedy 1962 Kefauver-Harris Amendments 1965 Declaration of Helsinki 1977 FDA Bioresearch Monitoring Program To better understand GCP, it helps to look at a brief history of the FDA and GCP. Biologics Act: Initial drug regulation passed to ensure purity and safety of serums, vaccines, and similar products used to prevent or treat diseases in humans. Came about as a result of the distribution of contaminated diphtheria antitoxin which caused the death of 12 children Food and Drug Act: Prohibited interstate commerce of adulterated and misbranded food and drugs. Established the US Pharmacopoeia and National Formulary as the official standards for drug manufacturing, quality, strength, and purity Elixir of Sulfanilamide Tragedy: Sulfanilamide containing the poisonous solvent diethylene glycol, kills 107 persons, many of whom are children, dramatizing the need to establish drug safety before marketing and to enact the pending food and drug law. Food, Drug, and Cosmetics Act: Requires new drugs to be shown safe before marketing-starting a new system of drug regulation. Thalidomide Tragedy: a new sleeping pill, is found to have caused birth defects in thousands of babies born in western Europe arouse public support for stronger drug regulation. Kefauver-Harris Amendments: passed to ensure drug efficacy and greater drug safety. For the first time, drug manufacturers are required to prove to FDA the effectiveness of their products before marketing them. Greater controls are placed on investigational drugs requiring submission of preclinical data to justify trials, reporting of clinical trial results, and informed consent. FDA Bioresearch Monitoring Program: AS a result of poor research practices, the FDA BIMO was created as an oversight group to ensure that research is being conducted to GCP/GLP standards. AKA FDA on-site Auditors.

20
History of GCP 1981 Informed Consent and IRB Regulations
1987 Regulations covering clinical investigator and sponsor obligations 1997 ICH GCP guidelines Computerized Systems Used in Clinical Investigations, May 2007 Protecting the Rights, Safety and Welfare of Study Subjects-Supervisory Responsibilities of Investigators, May 2007 FDAA, 2008 Informed Consent & IRB regulations: established the requirement for IRB review/approval of all human clinical studies. Established the requirement of obtaining and documenting informed consent. Declaration of Helsinki: World Medical Association declaration for ethical principles for medical research involving human subjects. States that the health of the patient will be the first concern of the physician. Research should be based on sound science and well designed protocols. ICH GCP Guidelines: International Conference on Harmonization Good Clinical Practice Guidelines established international standards. These will be reviewed in detail in this module.

21
Common Elements of GCP Human subject protection Informed Consent
Investigator obligations Sponsor obligations Monitor obligations Documentation/data handling/statistics Record keeping/archival Quality Assurance

22
Good Clinical Practice
Sponsor Regulatory Agency Clinical Investigator IRB/IEC Chart shows the interrelations of all the major people involved in a clinical trial. Provides a system of checks and balances to ensure that GCP is followed, human subjects are protected, and quality data is generated. Subject

23
Mission of FDA Good Clinical Practice Program
Focal point within FDA for Good Clinical Practice issues Coordinates FDA policies Contributes to leadership and direction through participation in FDA's Human Subject Protection/Bioresearch Monitoring Council Coordinates FDA's Bioresearch Monitoring program with respect to clinical trials, working together with FDA's Office of Regulatory Affairs (ORA) Contributes to international Good Clinical Practice harmonization activities Plans and conducts training and outreach programs Serves as a liaison with the HHS Office for Human Research Protection (OHRP) and other stakeholders of GCP
 Contributes to international Good Clinical Practice harmonization activities. Plans and conducts training and outreach programs. Serves as a liaison with the HHS Office for Human Research Protection (OHRP) and other stakeholders of GCP.")
24
Office for Good Clinical Practice (OGCP)
Established to be watchdog for GCP in clinical trials Committed to Quality Assurance in clinical trials “Quality assurance and quality improvement should become the prevailing themes in clinical research.” Quality Assurance: all those planned and systematic actions that are established to ensure that the trial is performed and the data are generated, documented (recorded), and reported in compliance with GCP and the applicable regulatory requirements. (definition from ICH GCP guidance)
, and reported in compliance with GCP and the applicable regulatory requirements. (definition from ICH GCP guidance)")
25
Office for Good Clinical Practice (OGCP)
In Clinical trials, you should strive for Building quality upfront Assuring Quality throughout Developing the capacity for continuous quality improvement now and in the future Quality Assurance is the cornerstone of success in GCP Quality is maintained via establishment of good processes for study conduct, good processes for data review and an overall process improvement initiative.

26
Office for Good Clinical Practice (OGCP)
Why do we need QA in Clinical Trials? More studies, more sites, and more volunteers at each site Expansion and fluidity of the clinical investigators pool New players in new roles (CROs, SMOs) New technologies (Electronic record keeping) More participation by vulnerable subjects Global expansion
 New technologies (Electronic record keeping) More participation by vulnerable subjects. Global expansion.")
27
What Defines U.S. GCPs? Food & Drug Administration’s Code of Federal regulations FDA Guidelines Other sources International Conference on Harmonisation Good Clinical Practice: Guideline (ICH/GCP) Hippocratic oath/Nurse Practice Act, etc. FDA’s Code of Federal Regulations: Part 11: Electronic Records and electronic signatures Part 50: Informed Consent Part 54: Financial Disclosure by clinical investigators Part 56: IRBs Part 312: Investigational New Drug Application Part 314 Applications for RDA approval to market a new drug FDA Guidelines: e.g. guideline on monitoring of clinical trials Other Sources: FDA Information Sheets IRB Clinical Investigator ICH GCP: This is what we will focus on for this discussion
 Hippocratic oath/Nurse Practice Act, etc. FDA’s Code of Federal Regulations: Part 11: Electronic Records and electronic signatures. Part 50: Informed Consent. Part 54: Financial Disclosure by clinical investigators. Part 56: IRBs. Part 312: Investigational New Drug Application. Part 314 Applications for RDA approval to market a new drug. FDA Guidelines: e.g. guideline on monitoring of clinical trials. Other Sources: FDA Information Sheets. IRB. Clinical Investigator. ICH GCP: This is what we will focus on for this discussion.")
28
FDA Regulations Electronic Records Protection of Human Subjects
21 CFR, part 11 Protection of Human Subjects 21 CFR, part 50 Financial Disclosure 21 CFR, part 54 Institutional Review Boards 21 CFR, part 56 Investigational New Drug Applications 21 CFR, part 312 Application for FDA Approval to Market a New Drug or an Antibiotic Drug 21 CFR, part 314 These are all links to the various FDA regulations. They are worth a brief exploration to better understand their content. The next several slides will discuss these regulations in greater detail.

29
21 CFR, Part 11

30
21 CFR, Part 11 Went into effect August 1997
Establishes the FDA’s requirements for electronic records and electronic signatures Applies to records in electronic format that are created, modified, maintained, archived, retrieved, or transmitted under any records requirements in FDA regulations.

31
21 CFR, Part 11 The regulation applies to source documents which are
Created in hard copy then entered into a computerized system Created by direct entry by a person into a computerized system Created automatically by a computerized system. Hard Copy examples: An ECG which is performed then the numerical values are entered into a computerized CRF Direct Entry: An adverse event which is recorded directly onto a computerized source document. It is not recorded on paper. In this case the source document. Automatic Entry: A blood pressure taken by a Dinamapp where the results are automatically downloaded onto an electronic source document for utlimate upload into an eCRF.

32
21 CFR, Part 50 Applies to all clinical investigations regulated by the FDA No investigator may involve a human being as a subject in research unless the investigator has obtained informed consent Details exceptions to informed consent Details 8 elements of informed consent IC must be documented using a written document of IC. Subpart D details the additional safeguards for children. Part 50 is related to Human Subject Protection for studies that are regulated by the FDA. This regulation does not apply to NIH studies. Those studies are governed by other legislation.

33
21 CFR, Part 54 Financial Disclosure
Purpose: To ensure that financial interests and arrangements of the clinical investigators are identified and disclosed. Applies to drugs, biologics and devices Intent is to make the agency aware of payment arrangements between commercial sponsors and investigators that could lead to inadvertent bias FDA will give closer scrutiny to studies where investigators have reportable financial interests or arrangements

34
21 CFR, Part 54 Financial Disclosure
Applicant must completely and accurately disclose or certify information concerning the financial interests of a clinical investigator who is not a full-time or part-time employee of the sponsor for each covered clinical study. Reporting Requirements Compensation affected by the outcome of clinical studies Significant equity interest in the sponsor of a covered study Proprietary interest in the tested product

35
21 CFR, Part 56 Investigational Review Boards
Composition, operation, and responsibility of an Institutional Review Board (IRB) that reviews clinical investigations regulated by the Food and Drug Administration Clinical investigation which must meet the requirements for submission to the FDA can not be initiated unless that investigation has been reviewed and approved by, and remains subject to continuing review by, an IRB Membership must have at least 5 members Diversity in race, gender One individual who is Scientist One individual who is a non-scientist One member not affiliated with the institution All IRBs are governed by this legislation.
 that reviews clinical investigations regulated by the Food and Drug Administration. Clinical investigation which must meet the requirements for submission to the FDA can not be initiated unless that investigation has been reviewed and approved by, and remains subject to continuing review by, an IRB. Membership must have at least 5 members. Diversity in race, gender. One individual who is Scientist. One individual who is a non-scientist. One member not affiliated with the institution. All IRBs are governed by this legislation.")
36
21 CFR, Part 56 Investigational Review Boards
Requires that IRB follows detailed written procedures Requires review of documents at convened meetings Allows for expedited review for situations with minimal risk

37
21 CFR, Part 312 Investigational New Drug Application
Details procedures and requirements for the use of investigational new drugs Allows you to ship drugs that have not been approved by the FDA for the purposes of conducting a clinical trial. Drug must be labeled with "Caution: New Drug--Limited by Federal (or United States) law to investigational use." FDA's primary objectives in reviewing an IND are, To assure the safety and rights of subjects, In Phase 2 and 3, to help assure that the quality of the scientific evaluation of drugs is adequate to permit an evaluation of the drug's effectiveness and safety. An IND is required for drugs that do not have FDA approval or for new formulations or indications.
 law to investigational use. FDA s primary objectives in reviewing an IND are, To assure the safety and rights of subjects, In Phase 2 and 3, to help assure that the quality of the scientific evaluation of drugs is adequate to permit an evaluation of the drug s effectiveness and safety. An IND is required for drugs that do not have FDA approval or for new formulations or indications.")
38
21 CFR, Part 312 Investigational New Drug Application
Sponsor submits IND when conducting a study with a drug that is regulated by part 312 Focus of the initial IND submission is on the general investigational plan and the protocols for specific human studies Amendments contain new or revised protocols build logically on previous submissions and include additional information Annual reports to the IND report the status of studies being conducted under the IND and update the general investigational plan for the coming year. Drugs regulated by part 312 include The clinical investigation of a drug product that is lawfully marketed in the United States is exempt from the requirements of this part if all the following apply: (i) The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication for use nor intended to be used to support any other significant change in the labeling for the drug; (ii) If the drug that is undergoing investigation is lawfully marketed as a prescription drug product, the investigation is not intended to support a significant change in the advertising for the product; (iii) The investigation does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product; (iv) The investigation is conducted in compliance with the requirements for institutional review set forth in part 56 and with the requirements for informed consent set forth in part 50; and (v) The investigation is conducted in compliance with the requirements of The amount of information on a particular drug that must be submitted in an IND depends upon such factors as the novelty of the drug, the extent to which it has been studied previously, the known or suspected risks, and the developmental phase of the drug.
 The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication for use nor intended to be used to support any other significant change in the labeling for the drug; (ii) If the drug that is undergoing investigation is lawfully marketed as a prescription drug product, the investigation is not intended to support a significant change in the advertising for the product; (iii) The investigation does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product; (iv) The investigation is conducted in compliance with the requirements for institutional review set forth in part 56 and with the requirements for informed consent set forth in part 50; and. (v) The investigation is conducted in compliance with the requirements of The amount of information on a particular drug that must be submitted in an IND depends upon such factors as the novelty of the drug, the extent to which it has been studied previously, the known or suspected risks, and the developmental phase of the drug.")
39
21 CFR, Part 314 Application to Market a New Drug
Applications to the FDA to review and approve a new drug to market. Establishes an efficient and thorough drug review process in order to: (a) Facilitate the approval of drugs shown to be safe and effective; and (b) ensure the disapproval of drugs not shown to be safe and effective. Also includes abbreviated applications
 Facilitate the approval of drugs shown to be safe and effective; and. (b) ensure the disapproval of drugs not shown to be safe and effective. Also includes abbreviated applications.")
40
FDA Guidelines FDA Information Sheets
FDA Guidelines for Monitoring of Clinical Investigations Guidance for IRBs, Investigators, and Sponsors FDA compliance Program Guidance Manuals FDA Compliance Policy Guidelines FDA Guidelines for the Preparation of IND Products

41
ICH/Good Clinical Practice
What is it? An international, ethical, and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects.

42
ICH/Good Clinical Practice
Goals: Identify and reduce differences in technical requirements for drug development among regulatory agencies Ensure worldwide acceptability of data Improve the quality of research Protection of research subjects

43
ICH/Good Clinical Practice
History: Initiated in 1990 with focus on Safety, Quality and Efficacy. Agreement between US, Europe and Japan to take action on harmonization Primarily concerned with studies conducted in United States, Japan, and the European Union As a result of the need for better quality in clinical trials the ICH GCP guideline was developed. 1997 Publication of Document E6 providing guidelines that describe the responsibilities and expectations of all participants in the conduct of clinical trials, Adopted by FDA as guidance document Prepared under the International Conference on Harmonization (ICH) ICH has many focuses addressing harmonization of drug trials including topics such as toxicology, manufacturing and pre-clinical data. One initiative of this large project is the E6 document that focuses on clinical study conduct. Although concerned primarily with the US, Japan, and EU, these guidelines should be used wherever research is conducted to generate clinical data that will be submitted to a regulatory authority.
 ICH has many focuses addressing harmonization of drug trials including topics such as toxicology, manufacturing and pre-clinical data. One initiative of this large project is the E6 document that focuses on clinical study conduct. Although concerned primarily with the US, Japan, and EU, these guidelines should be used wherever research is conducted to generate clinical data that will be submitted to a regulatory authority.")
44
International Conference on Harmonization (ICH)
E2D Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting E9 Statistical Principals for Clinical Trials E11 Clinical Investigation of Medicinal Products in Pediatric Population E14 The Clinical Evaluation of QT/QTc Interval Prolongation and Pro-arrhythmia Potential for Non-Anti-arrhythmic Drugs

45
ICH/Good Clinical Practice
General Points of Document E6 Protocol development Institutional Review Board (IRB)/Independent Ethics Committee (IEC) review and approval Informed consent processes Study conducted according to protocol Data recording, handling, storage Investigational drug accountability/control Adverse experience reporting Procedures to ensure quality of trial and data
/Independent Ethics Committee (IEC) review and approval. Informed consent processes. Study conducted according to protocol. Data recording, handling, storage. Investigational drug accountability/control. Adverse experience reporting. Procedures to ensure quality of trial and data.")
46
ICH/Good Clinical Practice
ICH Guidelines cover the following topics: Institutional Review Board (IRB)/Independent Ethics Committee (IEC) Investigator Sponsor Protocol Investigator: includes obtaining an informed consent Sponsor: includes trial monitoring and audit
/Independent Ethics Committee (IEC) Investigator. Sponsor. Protocol. Investigator: includes obtaining an informed consent. Sponsor: includes trial monitoring and audit.")
47
ICH/Good Clinical Practice
Investigator’s Brochure (IB) Essential documents for the conduct of a clinical trial Investigator’s Brochure is information on studies, both clinical and pre-clinical, conducted to date. It is expected that the Investigator and anyone involved in the preparation of clinical supply is familiar with the IB.
 Essential documents for the conduct of a clinical trial. Investigator’s Brochure is information on studies, both clinical and pre-clinical, conducted to date. It is expected that the Investigator and anyone involved in the preparation of clinical supply is familiar with the IB.")
48
Institutional Review Board (IRB)/Independent Ethics Committee (IEC)
Primary function is to safeguard the rights, safety, and well-being of all trial subjects, especially vulnerable subjects Local IRB vs Central IRB The ICH GCP guidelines include everything from the composition of the IRB to what documents need to be review and approved. The IRB will be discussed in detail in another training module. Independent Ethics Committee is European version.

49
Investigator Responsibilities – Ethical Principles
Medical management of study subjects Qualifications Adequate Resources Medical Care of Trial Subjects Communication with IRB/IEC Compliance with Protocol Investigational Product Informed Consent Patient records/case report forms

50
Investigator Responsibilities
Records and Reports Progress Reports Safety Reporting Premature termination/suspension of trial Final Report Records & Reports: Investigator is responsible for the accuracy, completeness, legibility, and timeliness of the data in the CRFs. Source documentation and CRFs will be covered in more detail in another module. Progress Reports: The investigator is responsible for submitting written summaries of the trial to the IRB annually as well as reports to the sponsor. Safety Reporting: The Investigator is responsible for serious AE reporting. AEs will be covered in more detail in another module. Premature Termination/Suspension of Trial: If a trail is terminated early or suspended, the investigator is responsible for notifying the subjects, assuring appropriate therapy and follow up for the subjects, and where applicable, informing the regulatory authority. Final Report: At trial end, the investigator is responsible for providing the sponsor will all required reports, and providing the IRB with a summary of the trials outcome.

51
Sponsor Responsibilities
Medical expertise – Investigator’s Brochure Study design Trial management, Data management Data Handling, Recordkeeping, and Independent Data Monitoring Committee Supervise conduct of study Data handling and verification Trial Management, Data Handling: Use qualified individuals to supervise the overall conduct of the trail, to handle the data, to verify the data, to conduct the statistical analyses, and to prepare the trial reports. When using electronic data systems: the sponsor/CRO must ensure/document the system has been validated, maintain SOPs for it, ensure that it has an audit trail for any data changes, a security system to prevent unauthorized access, adequate backup, and safeguard the blinding of the study. Investigator Selection: Sponsor is responsible for selecting the investigator/institution. Confirmation of Review by IRB/IEC: Sponsor should obtain from investigator documented IRB/IEC approval. Safety Information: Sponsor is responsible for the ongoing safety eval of the drug and notifying all involved investigators of anything that could adversely affect the safety of the subjects.

52
Sponsor Responsibilities
Investigator selection Confirmation of review by IRB/IEC Safety reporting Record management

53
Sponsor Responsibilities
Audit Evaluate trial conduct is in compliance with protocol, GCP and SOP Monitoring Purpose: insure rights of subjects are protected, data are accurate, complete and verifiable, trial conducted to GCP Selection/Qualifications of Monitor Audit: The purpose of the audit is to evaluate the trial conduct and compliance with the protocol, SOPs, GCPs, and applicable regulatory requirements. An audit may be done as part of a quality assurance program. It can be conducted by an group internal to the site, internal to the sponsor or a regulatory body such as the FDA. Monitoring: Serves as sponsor representative to the site, ensuring that the study was conducted in accordance with the protocol and GCP Purpose: Purpose of monitoring is to make sure the rights of the subjects are protected, the reported data are accurate, complete, and verifiable from source documents, and the trial was conducted in compliance with the protocol, GCP, and regulatory requirements. Selection/Qualifications: The sponsor is responsible for appointing the monitor. The monitor needs to be familiar with the investigational product, the protocol, the consent form, and any other written information.

54
Sponsor Responsibilities
Quality Assurance/Quality Control SOPs regarding trial conduct Registration of clinical trials Contract Research Organization Sponsor may transfer all trial responsibilities to a CRO CRO must implement QA/QC All sponsor responsibilities transfer to CRO QA/QC: Responsible for implementing and maintaining qa/qc systems with written SOPs to ensure that trials are conducted in compliance with GCP CRO: The Sponsor may transfer any or all of his duties and functions to a CRO but retains the ultimate responsibility for them. The CRO should implement qa/qc. All responsibilities referred to as the sponsors should also apply to the CRO.

55
Monitoring Responsibilities
Main line of communication Verify investigator qualifications Monitor handling of investigational product Verify adherence to protocol Informed consent Manage study regulatory documents on behalf of the sponsor Ensure site training is provided Provide Monitoring Report Responsibilities: In general, there needs to be on-site monitoring before, during and after the trial. The monitor should: Act as the main line of communication between the sponsor and the investigator Verify that the investigator has adequate qualifications and resources Verify for the investigational product that the drug has been stored and dispensed appropriately, and that unused supplies have been handled appropriately Verify that the protocol has been followed Verify that written consent has been obtained Ensure that the investigator receives the current IB, documents, and supplies to conduct the trial appropriately Ensure that the investigator and staff are properly educated about the trial Verify that only eligible subjects are being enrolled Report the subject recruitment rate Verify that source data/documents are accurate and complete Verify that the investigator provides all the required reports and submissions Checks the accuracy and completeness of the CRF entries, source documents and other trial related records against each other. Specifically Data on CRFs are consistent with the source docs Any dose or therapy modifications are well documented AEs, Concomitant medications, and inter current illnesses are reported as per protocol Missed procedures, visits are recorded on the CRFs Withdrawals or dropouts are reported and explained on the CRFs Informing the investigator of any CRF entry error, omission or illegibility. Determining whether all Ads are appropriated reported within the time periods as per GCP, protocol, and regulatory requirements Communicating deviations from the protocol, SOPs, and GCP to the investigator and taking appropriate action to prevent recurrences Monitoring Report: must submit a written report to the sponsor after each trial-site visit or trial-related communication all findings.

56
Protocol and Amendments
Should include General Information Background Information Trial Objectives and Purpose Trial Design Selection/Withdrawal of subjects Treatment of Subjects Assessment of Efficacy Assessment of Safety Statistics General Information: Protocol title, number, date as well as demographic information of the sponsor and investigators Background Information: name and description of the investigational product, summary on non-clinical and clinical findings, summary of know and potential risks and benefits to the subjects, description of the population to be studies. Trial Objectives/Purpose: a detailed description of the objectives and purpose of the trial. Trial Design: specific statement of the primary and secondary endpoints, description of the design of the trial, ways to minimize or avoid bias (e.g. blinding, randomization), description of the dosage and regimen, and expected duration of participation. Selection and Withdrawal of Subjects: Inclusion/exclusion criteria, and withdrawal criteria Treatment of Subjects: treatment to be administered, dosing schedule, route of administration, treatment period, medications permitted and not permitted during trial, and procedures for monitoring compliance. Assessment of Efficacy: efficacy parameters and methods and timing for assessing, recording, and analyzing them. Assessment of Safety: safety parameters, and methods and timing for assessing, recoding, and analyzing. Procedures for eliciting reports of and for recording and reporting AEs and intercurrent illnesses. Statistics: number of subjects planned to be enrolled and reason for sample size, level of significance. Criteria for the termination of the trial.
, description of the dosage and regimen, and expected duration of participation. Selection and Withdrawal of Subjects: Inclusion/exclusion criteria, and withdrawal criteria. Treatment of Subjects: treatment to be administered, dosing schedule, route of administration, treatment period, medications permitted and not permitted during trial, and procedures for monitoring compliance. Assessment of Efficacy: efficacy parameters and methods and timing for assessing, recording, and analyzing them. Assessment of Safety: safety parameters, and methods and timing for assessing, recoding, and analyzing. Procedures for eliciting reports of and for recording and reporting AEs and intercurrent illnesses. Statistics: number of subjects planned to be enrolled and reason for sample size, level of significance. Criteria for the termination of the trial.")
57
Investigator’s Brochure
Compilation of the clinical and nonclinical data on the investigational drug. Needs to reviewed at least annually and revised as necessary As per GCP, relevant new information may be so important that it should be communicated to the investigators before it is included in a revised IB IND safety updates Copy provided to IRB. Copy of the IB should be given to the Investigator and subsequently reviewed by the investigator and also submitted to IRB.

58
KEYS to GCP Follow If it’s not documented, it didn’t happen!
The regulations The protocol The SOPs Guidelines If it’s not documented, it didn’t happen! Must be able to recreate CRF data from source documents

59
Links FDA 21 CFR, part 54 FDA 21 CFR, part 56 FDA 21 CFR, part 312
E6 Consolidated Guidance for Good Clinical Practice FDA 21 CFR, Part 50 FDA 21 CFR, part 54 FDA 21 CFR, part 56 FDA 21 CFR, part 312 FDA 21 CFR, part 314 NIH

60
Role of the Principal Investigator
care Role of the Principal Investigator community Donna W. Dorozinsky, RN, MSN, CCRC research teach

61
Objectives Describe the Investigator responsibilities as defined by the FDA and ICH Discuss the practical elements necessary to remain in regulatory and ethical compliance with all aspects of the clinical trial process Discuss the additional responsibilities of the Investigator in Investigator initiated research.

62
GCP and the and the Investigator
Golden Standards To ensure the integrity of clinical research data To provide adequate safeguards to ensure the protection of human subjects To ensure that the highest standard of ethical conduct is preserved To address all applicable regulatory requirements

63
Expectations What does the FDA expect of you?
Manage the clinical trial Conduct the protocol Collect quality data Investigational article accountability

64
Expectations What does the FDA expect of you?
Manage the clinical trial Responsibility, authority, accountability, delegation, and documentation Conduct the protocol Who did what? Who obtained informed consent? Who determined participant eligibility? Who corresponded with the IRB? Who identified, reported and followed-up on AEs?

65
Expectations What does the FDA expect of you? Quality data
Who maintained study files? Who collected the data? Who completed the case report forms? What source documents are used to validate and support data submitted to FDA? Investigational article accountability Where was the investigational drug stored? Who dispensed the drug? Who can account for the investigational drug?

66
Statement of Investigator
Your commitments Form FDA 1572

68
Please DO NOT RETURN this application to this address.
8. ATTACH THE FOLLOWING CLINICAL PROTOCOL INFORMATION: FOR PHASE 1 INVESTIGATIONS, A GENERAL OUTLINE OF THE PLANNED INVESTIGATION INCLUDING THE ESTIMATED DURATION OF THE STUDY AND THE MAXIMUM NUMBER OF SUBJECTS THAT WILL BE INVOLVED. FOR PHASE 2 OR 3 INVESTIGATIONS, AN OUTLINE OF THE STUDY PROTOCOL INCLUDING AN APPROXIMATION OF THE NUMBER OF SUBJECTS TO BE TREATED WITH THE DRUG AND THE NUMBER TO BE EMPLOYED AS CONTROLS, IF ANY; THE CLINICAL USES TO BE INVESTIGATED; CHARACTERISTICS OF SUBJECTS BY AGE, SEX, AND CONDITION; THE KIND OF CLINICAL OBSERVATIONS AND LABORATORY TESTS TO BE CONDUCTED; THE ESTIMATED DURATION OF THE STUDY; AND COPIES OR A DESCRIPTION OF CASE REPORT FORMS TO BE USED. 9. COMMITMENTS: I agree to conduct the study(ies) in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, rights, or welfare of subjects. I agree to personally conduct or supervise the described investigation(s). I agree to inform any patients, or any persons used as controls, that the drugs are being used for investigational purposes and I will ensure that the requirements relating to obtaining informed consent in 21 CFR Part 50 and institutional review board (IRB) review and approval in 21 CFR Part 56 are met. I agree to report to the sponsor adverse experiences that occur in the course of the investigation(s) in accordance with 21 CFR I have read and understand the information in the investigator’s brochure, including the potential risks and side effects of the drug. I agree to ensure that all associates, colleagues, and employees assisting in the conduct of the study(ies) are informed about their obligations in meeting the above commitments. I agree to maintain adequate and accurate records in accordance with 21 CFR and to make those records available for inspection in accordance with 21 CFR I will ensure that an IRB that complies with the requirements of 21 CFR Part 56 will be responsible for the initial and continuing review and approval of the clinical investigation. I also agree to promptly report to the IRB all changes in the research activity and all unanticipated problems involving risks to human subjects or others. Additionally, I will not make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to human subjects. I agree to comply with all other requirements regarding the obligations of clinical investigators and all other pertinent requirements in 21 CFR Part 312. INSTRUCTIONS FOR COMPLETING FORM FDA 1572 STATEMENT OF INVESTIGATOR: 1. Complete all sections. Attach a separate page if additional space is needed. 2. Attach curriculum vitae or other statement of qualifications as described in Section 2. 3. Attach protocol outline as described in Section 8. 4. Sign and date below. 5. FORWARD THE COMPLETED FORM AND ATTACHMENTS TO THE SPONSOR. The sponsor will incorporate this information along with other technical data into an Investigational New Drug Application (IND). 10. SIGNATURE OF INVESTIGATOR 11. DATE (WARNING: A willfully false statement is a criminal offense. U.S.C. Title 18, Sec ) Public reporting burden for this collection of information is estimated to average 100 hours per response, including the time for reviewing instructions, searching existing data sources, gathering and maintaining the data needed, and completing reviewing the collection of information. Send comments regarding this burden estimate or any other aspect of this collection of information, including suggestions for reducing this burden to: Food and Drug Administration CBER (HFM-99) 1401 Rockville Pike Rockville, MD CDER (HFD-94) 12229 Wilkins Avenue Rockville, MD 20852 "An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number." Please DO NOT RETURN this application to this address.
 in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the sponsor, except when necessary to protect the safety, rights, or welfare of subjects. I agree to personally conduct or supervise the described investigation(s). I agree to inform any patients, or any persons used as controls, that the drugs are being used for investigational purposes and I will ensure that the requirements relating to obtaining informed consent in 21 CFR Part 50 and institutional review board (IRB) review and approval in 21 CFR Part 56 are met. I agree to report to the sponsor adverse experiences that occur in the course of the investigation(s) in accordance with 21 CFR I have read and understand the information in the investigator’s brochure, including the potential risks and side effects of the drug. I agree to ensure that all associates, colleagues, and employees assisting in the conduct of the study(ies) are informed about their obligations in meeting the above commitments. I agree to maintain adequate and accurate records in accordance with 21 CFR and to make those records available for inspection in accordance with 21 CFR I will ensure that an IRB that complies with the requirements of 21 CFR Part 56 will be responsible for the initial and continuing review and approval of the clinical investigation. I also agree to promptly report to the IRB all changes in the research activity and all unanticipated problems involving risks to human subjects or others. Additionally, I will not make any changes in the research without IRB approval, except where necessary to eliminate apparent immediate hazards to human subjects. I agree to comply with all other requirements regarding the obligations of clinical investigators and all other pertinent requirements in 21 CFR Part 312. INSTRUCTIONS FOR COMPLETING FORM FDA 1572 STATEMENT OF INVESTIGATOR: 1. Complete all sections. Attach a separate page if additional space is needed. 2. Attach curriculum vitae or other statement of qualifications as described in Section Attach protocol outline as described in Section Sign and date below. 5. FORWARD THE COMPLETED FORM AND ATTACHMENTS TO THE SPONSOR. The sponsor will incorporate this information along with other technical data into an Investigational New Drug Application (IND). 10. SIGNATURE OF INVESTIGATOR. 11. DATE. (WARNING: A willfully false statement is a criminal offense. U.S.C. Title 18, Sec ) Public reporting burden for this collection of information is estimated to average 100 hours per response, including the time for reviewing instructions, searching existing data sources, gathering and maintaining the data needed, and completing reviewing the collection of information. Send comments. regarding this burden estimate or any other aspect of this collection of information, including suggestions for reducing this burden to: Food and Drug Administration. CBER (HFM-99) 1401 Rockville Pike. Rockville, MD CDER (HFD-94) Wilkins Avenue. Rockville, MD An agency may not conduct or sponsor, and a. person is not required to respond to, a. collection of information unless it displays a. currently valid OMB control number. Please DO NOT RETURN this application to this address.")
69
Investigator Responsibilities FDA Form 1572
Protocol compliance. Agree to… conduct of the trial according to the Protocol. Personally supervise the study Inform subjects of drugs that are being used for investigational purposes Ensure all clinic staff fully understand and follow the protocol Implement changes to the protocol only after written consent of sponsor, IRB and regulatory agencies. Report AEs Read and understand the IB Maintain records Comply with 21 CFR Parts 56 and 312 I.

70
Investigator Responsibilities
Medical management of trial participants To provide information to the participant regarding nature of the investigation To provide adequate medical care for the participant during the trial To ensure welfare and safety of the participant during the trial To provide appropriate medical care and follow-up procedures at the conclusion of the trial These are not ordinary patients

71
Translation Into Practice
Answer subject questions Medical care takes priority over the protocol Safety comes first Follow events to resolution Communication with PCP Subject access to PI

72
Investigator Responsibilities
Staff and Facilities To ensure that there is adequate staff to support the study, that they are familiar with the trial protocol and the standards of Good Clinical Practices To ensure adequate facilities and equipment To use certified laboratories with validated assay methodology

73
Translation Into Practice
Study Coordinator Facilities and Equipment Secured storage of IP and study files Evidence of equipment maintenance Validated assays

74
Investigator Responsibilities
Adequate Resources Potential for recruiting adequate number of qualified subjects. Sufficient time to properly conduct study Adequate staff and resources Ensure all staff involved in conduct of study have been properly trained

75
Translation Into Practice
Available qualifying patients Research is time consuming Are your coordinators carrying too many studies? If your subjects need eye examinations do you have arrangements with an ophthalmologist?

76
Investigator Responsibilities
Compliance with local regulations and institutional requirements To obtain the review/approval of the IRB To provide periodic study status reports to the IRB (at least annually) To promptly inform the IRB / DSMB of significant safety issues (SAEs from site, IND Safety Reports for program) Informed Consent
 To promptly inform the IRB / DSMB of significant safety issues (SAEs from site, IND Safety Reports for program) Informed Consent.")
77
Translation Into Practice
Communication with a duly constituted IRB Local vs. Commercial Annual update Communicating IND safety updates to the IRB Evaluating IND safety updates to determine need for adjustments to ICF Process for identifying and reporting SAEs Process for obtaining informed consent

78
Investigator Responsibilities
Control of investigational drugs, vaccines, or devices (article) To provide accountability system to address receipt, distribution, and return of all investigational supplies To ensure that supplies are properly stored and accessible only to designated study staff To ensure that investigational materials are used only according to protocol To ensure that randomization procedures exist along with process for breaking the blind May be delegated to a qualified individual Explain correct use to the subject Procedures for randomization and blinding
 To provide accountability system to address receipt, distribution, and return of all investigational supplies. To ensure that supplies are properly stored and accessible only to designated study staff. To ensure that investigational materials are used only according to protocol. To ensure that randomization procedures exist along with process for breaking the blind. May be delegated to a qualified individual. Explain correct use to the subject. Procedures for randomization and blinding.")
79
Translation Into Practice
Process for receipt storage and return Secure IP storage area that is monitored for temperature Drug Accountability performed each time drug is dispensed Establish procedure for maintaining randomization and breaking the blind

80
Investigator Responsibilities
D O C U M E N T A I Patient records/case report forms To provide complete and accurate data to the sponsor To maintain patient records to include history, prescribed medication and investigational product(s), measurements, exams, evaluations and adverse events To apply corrections to clinical research data according to principles of good research practice (i.e., single-line delete, date and initial) Correlation between the CRF and the source document
, measurements, exams, evaluations and adverse events. To apply corrections to clinical research data according to principles of good research practice (i.e., single-line delete, date and initial) Correlation between the CRF and the source document.")
81
Translation Into Practice
D O C U M E N T A I Patient records/case report forms Complete and accurate data to the sponsor Patient records that include history, measurements, exams, evaluations and adverse events Access to in-patient records QC of CRFs to source documents

82
Translation Into Practice
Regulatory Binder IRB Communications Protocol, ICF, Amendments IB FDA Form 1572 Financial Disclosure CVs Site Signature Log Delegation of Authority Log Study Communications Screening Log Laboratory Reference Ranges Site Visit Log IP Accountability

83
Translation Into Practice
Archive Essential Study Documents at end of study Should be easy to retrieve documents If you move your location, notify the sponsor

84
Investigator Responsibilities
Safety Reporting SAEs reported immediately to the sponsor AE or laboratory abnormalities reported to the sponsor within the agreed timeline. Report deaths and all associated documentation to sponsor and IRB.

85
Translation Into Practice
Assess for AEs at each visit IB is the driver for determining SAEs Key is “unexpected” Report all adverse events – you never know! All clinically significant laboratory or diagnostic results are AEs Functioning under an assurance Unexpected events Unexpected problems

86
Investigator Responsibilities
Compliance with the Protocol Conduct the study as described in the protocol No deviations without agreement from sponsor and IRB Documentation of any unintentional deviations Deviation for immediate hazard

87
Translation Into Practice
Follow the protocol as it is written Recipe versus Contract Defined process for training staff involved in conduct of the trial Do not implement a change in the protocol until you have received written IRB approval. Revision to ICF

88
Investigator Responsibilities
Informed consent Comply with all regulatory requirements and adhere to GCP Revise ICF when new information becomes available There should be no coercion of the subject to participate Use non-technical language Provide ample time for consenting Copy of the signed and dated ICF

89
High Risk to Delegate Approval for study participation
Assignment of causality for adverse events (AE) Physical examinations Final review of CRFs
 Physical examinations. Final review of CRFs.")
90
Clarification of FDA expectations:
Protecting the Rights, Safety, and Welfare of Study Subjects – Supervisory Responsibilities of Investigators Draft FDA Guidance, May 2007 Responsibilities related to human subject protection and data integrity. Clarification of FDA expectations: To supervise a clinical study where some study tasks are delegated To protect rights, safety and welfare of study subjects.

91
Clinical Trials of Drugs
Clarification of Investigator Responsibilities Supervision of Conduct of the Study Protecting the Rights, Safety and Welfare of Study Subjects

92
Supervision of Conduct of Study
Appropriate delegation of tasks Ensure that individuals to whom a task is delegated is qualified to perform the task. Generally related to tasks that are clinical or medical in nature. Historically in appropriate delegation of tasks.

93
Supervision of Conduct of Study
Define adequate training General familiarity with the protocol Specific understanding of the details related to the tasks they will be performing Awareness of regulatory requirements and acceptable standards Competency Informed of changes to protocol

94
Supervision of Conduct of Study
Adequate Supervision Routine meetings with staff to review trial progress and update staff Routine meetings with the sponsor’s monitors Procedure for correcting problems identified by study personnel. documenting the performance of delegated tasks in a satisfactory manner. ensuring study is conducted in accordance with 21 CFR, Part 50. Ensuring that information in source documentation matches CRFs. dealing with data queries and discrepancies identified by the study monitor. Ensuring staff comply with protocol, AE assessment and reporting and other medical issues.

95
Supervision of Conduct of Study
Oversight of other parties involved in the conduct of the study. Study staff not in direct employ of the investigator. Parties other than study staff (clinical laboratories

96
Protecting the Rights, Safety and Welfare of Study Subjects
Provide a reasonable standard of medical care. Reasonable access to medical care by being available to subjects during the conduct of the trial at their site. Seek to minimize protocol violations which may be considered a failure to protect the rights, safety and welfare of subjects Access to medical care: If investigator is not going to be available for an extended period, then clinical responsibility for study subjects should be delegated to a specific qualified physician who will be readily available to subjects. Document this availability in a 1572 or investigator agreement (with notification to IRB). In the clinical investigator is a non-physician, then adequate arrangements should be made for medical coverage.
. In the clinical investigator is a non-physician, then adequate arrangements should be made for medical coverage.")
97
Investigator Initiated Research
Reasons Investigators choose to act as a sponsor Scientific interest in a drug or product An opportunity to contribute to clinical knowledge A potential for publication of study results

98
Investigator Initiated Research
Responsibilities Writing the protocol and designing the Case Report Form Monitoring the study and reviewing the source documents Drug accountability Submitting safety reports to the FDA Complying with all applicable FDA regulations IND submission

99
Questions ???

100
Human Subject Protection
care Human Subject Protection community Donna W. Dorozinsky, RN, MSN, CCRC research teach

101
Course Objectives Discuss the history of human subject protection and the historical events impacting today’s current regulations. Identify the 8 elements of informed consent. Discuss the general requirements in obtaining informed consent. Discuss the IRB requirements and the contents of 21 CFR, Part 56. Discuss the HIPAA requirements as they relate to privacy in clinical research.

102
Human Subject Protection
History of Human Subject Protection Declaration of Helsinki Belmont Report Informed Consent Institutional Review Boards

103
Historical Abuse of Subjects
1932: Tuskegee syphilis study Designed to determine the natural history of untreated syphilis 400 black men with syphilis were recruited without informed consent Syphilis left untreated to determine course of disease 100 men died, 40 wives infected, 19 children contracted disease at birth World War II Appalling experiments conducted on imprisoned ethnic groups by physicians Tuskegee: first account of this was reviewed in In 1997 the US made a formal apology to remaining survivors and families.

104
Human Subject Protection
1947: Nuremberg Code 1962: Kefauver-Harris Amendment 1964: Declaration of Helsinki 1967: FDA required informed consent to be obtained in writing 1974: National Research Act 1979: Belmont report 1981: Congress enacted 3 statutes that govern human subject protection Nuremberg Code: Response to WWII atrocities. Was the first declaration of medical ethics. Required voluntary consent to be given by subjected participating in medical research. Kefauver-Harris Amendment: Required informed consent Declaration of Helsinki: Recommendations guiding physicians in biomedical research involving human subjects. Will be explored in more depth 1967: FDA required that the consent process and required consent be obtained in writing for early stages of research National Research Act: Mandated the establishment of IRBs to review all federally funded human research. Created the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. Belmont Report: Objective was to provide an analytical framework that will guide the resolution of ethical problems arising from research involving human subjects. Will be explored in more detail. 1981: FDA regulation 21 CFR part 50, PHS regulation 45 CFR part 46 both titled Protection of Human Subjects. Also FDA regulation 21 CRF part 56 titled Institutional Review Boards. These are the governing documents today.

105
Declaration of Helsinki
Research must conform to generally accepted scientific principles and should be based on adequately performed laboratory and animal experiments. Follow a protocol which is reviewed by a “specially appointed independent committee for consideration, comment, and guidance” e.g. IRB Conducted only by scientifically qualified persons; supervised by clinically competent medical person Importance of the objective is in proportion to the risk to the subject Predictable risks outweigh foreseeable benefits Respect the privacy of the subject and safeguard his/her integrity Adopted in 1964, amended in 1975 & 1983 by the World Medical Assembly, multiple amendments since. Placebo requirements. Gave recommendations to serve as a guide to every physician in biomedical research involving human subjects. These are only a guide.

106
Declaration of Helsinki
Each subject must give informed consent indicating they he/she is adequately informed of the study, benefits, hazards; they must be informed that he/she can decide not to participate or to withdraw from the study If subject is incompetent, then informed consent must be obtained from the legal guardian. If the subject is a minor who can give consent, this should be obtained in addition to the legal guardian’s.

107
Belmont Report Objective is to provide an analytical framework that will guide the resolution of ethical problems arising from research involving human subjects. Published in April Written by the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. This was a select group of 11 American men and women. Their assignment from congress was to “identify the basic ethical principles that should underlie the conduct of biomedical and behavioral research involving human subjects and to develop guidelines which should be followed to assure that such research is conducted in accordance with those principles”. These have never been formally adopted but have become the primary ethical framework for protecting human research subjects in the United States. This became the basis for future DHHS laws.

108
Belmont Report Covers 3 topics
Boundaries between practice and research Basic ethical principles Applications

109
Belmont Report Boundaries between practice and research
Practice: interventions to enhance the well-being of a patient with a reasonable expectation of success. To provide diagnosis and preventive treatment or therapy. Research: Activity designed to test a hypothesis, draw conclusions, and contribute to general knowledge. Formal protocol with an objective and set of procedures. Benefits are not always known. May be carried on together when the research is to evaluate the safety/efficacy of a new therapy. If there is any element of research, it needs to undergo review for the protection of human subjects, i.e. IRB review. During informed consent it is essential that the subject know which activities are research and which are standard practice.

110
Belmont Report Basic Ethical Principles
Respect for Persons: Individuals should be treated as autonomous agents Persons with diminished autonomy are entitled to protection Subjects must enter into the research voluntarily and with adequate information. Beneficence An obligation to improve a persons’ well being. Do not harm Maximize possible benefits Minimize possible risks 1. Autonomous person: someone capable of deliberation about personal goals and acting upon their decisions. Diminished autonomy: includes those people with diminished mental capacity, minors, prisoners. Hippocratic oath: to help or at least to do no harm. Must decide when it is proper to pursue benefits of research despite the risks involved and when should the benefits be foregone because of risks to the subjects.

111
Belmont Report Applications
Informed consent: People should have the opportunity to choose what shall or shall not happen to them. Contains 3 elements: information, comprehension, voluntariness Information: subject must understand that research is neo necessary for their well being nor are the effects of the research fully known. Comprehension: The information provided to the subject must be in a language and at a level that he/she can understand. Investigators are responsible for making sure that a subject understands the information. Voluntariness: A consent is valid only if it is voluntarily given which means it is free of coercion and undue influence. The report singles out the following three activities: Informed consent, risk/benefit assessment, subject selection Informed consent: Information: specific items to include: research procedure, purposes, risks, benefits, alternative procedures, statement offering the subject the chance to ask questions and to withdraw from the research at any time. Comprehension: The manner and context that the information is presented is as important as the information itself. When the study risks are more serious, the obligation to make sure the subject understands the information is even more important. In some cases it may be suitable to give the subject an oral or written test of comprehension. Voluntariness: Coercion is the overt threat of harm in order to obtain compliance. Undue influence is an offer of excessive, unwarranted, inappropriate or improper reward in order to gain compliance. This could be excessive payment in a study.

112
Belmont Report Applications
Assessment of Risks and Benefits: Risk: the possibility that harm may occur Benefit: something positive related to well being. Risks to the subject should be outweighed by the sum of anticipated benefits to the subject and society Selection of Subjects: Two levels of justice: individual and societal Individual justice: Investigator must demonstrate fair procedures in selecting the subjects. Societal justice: fair and equitable selection of subjects across economic, ethnic, and gender classes.

113
Informed Consent

114
Informed Consent Governing documents:
FDA 21 CFR 50: Protection of Human subjects HHS 45 CFR 46: Protection of Human Subjects FDA Information Sheet: Informed Consent Regulations FDA Information Sheet: A Guide To Informed Consent Documents

115
Informed Consent “Informed consent is more than just a signature on a form, it is a process of information exchange that includes, recruitment materials, written materials, verbal instructions, question/answer sessions, and measure of subject understanding.” Quote from FDA information sheet “A Guide to Informed Consent Documents”

116
Informed Consent “The consent document should be the basis for a meaningful exchange between the investigator and the subject.” Quote from FDA Information sheet: “A Guide to Informed Consent” the consent process

117
Ethical Standards of Informed Consent
One of the biggest obligations facing everyone involved in developing new medicines and medical devices that use the human research subject is the consistent requirement of Respect, compassion, understanding JAMA, 5/24/00, Vol 283, No. 20 “What makes Clinical Research Ethical?

118
Informed Consent Informed Consent Definition
A process by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed of all aspects of the trial that are relevant to the subject’s decision to participate. Informed consent is documented by means of a written, signed, and dated informed consent form. Definition from ICH GCP E6 document

119
Informed Consent Informed consent documents must meet the requirements of 21 CRF and contain the information required by 21 CRF IRBs have the ultimate authority for ensuring the adequacy of the information in the informed consent document. IRBs: Many have standard language and/or a standard format to be used in portions of the IC documents. Typical areas with standard language include confidentiality, compensation, answers to questions, and the voluntary nature of participation.

120
Informed Consent General Requirements for Informed Consent:
No investigator may involve a human being as a subject in research unless informed consent is obtained. Sufficient opportunity must be give to the subject to decide whether or not to participate

121
Informed Consent General Requirements for Informed Consent:
Must minimize the possibility of coercion or undue influence No informed consent may include any exculpatory language (release of responsibility). Exculpatory language: waive or appear to waive any of the subject’s rights. Releases or appears to release the investigator, the sponsor, the institution, from liability for negligence.
. Exculpatory language: waive or appear to waive any of the subject’s rights. Releases or appears to release the investigator, the sponsor, the institution, from liability for negligence.")
122
Informed Consent Informed consent must be understandable
Technical and scientific terms must be adequately explained or simpler terms substituted. More understandable if the subject is referred to as “you” and the investigator as “I/we” Subjects should not be asked to certify that they “fully understand” the study. Subjects may state that they understand the information in the IC document or the information presented to them. They cannot judge whether the information in the document is complete.

123
Informed Consent Informed consent must be understandable
Consent must not imply or state that the study is approved by the FDA If subject population includes non-English specking subjects, a translated consent document should be prepared Studies specifically are not approved by the FDA. FDA approves an IND but not each particular study. IRB should review translated document to be sure that it is accurate. If a language barrier does exist, the investigator should consider the ethical implications of enrolling the subject. They may not clearly understand the information presented, therefore, consent will not truly be informed.

124
Informed Consent Informed consent must be understandable
A person who understands English, but does not read and write can be consented by their “making their mark” on the document. When consenting older children, it is recommended that there be two forms: an informed consent for the parent/guardian to sign as well as an assent document for the child to sign. To consent an illiterate subject, the document should be read to the subject followed by the usual consent discussion. An impartial third party should witness the consent process and sign the consent document. It is recommended that the procedure is also videotaped. An assent document is defined as “a child’s affirmative agreement to participate in research.”

125
Can they read the consent form?
48% Of American adults Have low literacy skills

126
Who are we protecting? Job applicants tested in >33% lacked reading and mathematics skills for employment! Question? Do we expect these people to read and understand the consent form?

127
Elements of Informed Consent
Statement that the study involves research, explanation of the purposes of the research, expected duration of the subject’s participation, description of the procedures to be followed, identification of any procedures which are experimental. Elements are specified in 21 CRF 50.25 Statement that the study involves research is important to clarify between the physician/patient relationship which is different than the investigator/subject relationship. Research procedures should be explained using simple language: e.g. randomization, placebo control Consent should include statement that the safety of the investigational product will be evaluated. It should not imply that the test articles are safe or that safety has already been established.

128
Elements of Informed Consent
Description of any reasonably foreseeable risks or discomforts to the subject. Description of any benefits to the subject or to others which may reasonably be expected from the research. Disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject. Risks of any procedures related solely to the research should be included in the document. Risks of the test article should be based on information in the protocol, IB, package labeling, and previous research. Clear/not overstated. If not direct benefit, that should be stated. Subjects should be aware of the full range of options available.

129
Elements of Informed Consent
Statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained and that notes the possibility that the FDA may inspect the records. For research involving more than minimal risk, an explanations to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs. Should be informed of the extent that the institution intends to maintain confidentiality. If sponsor may have access to records that should be included. Absolute protection of confidentiality by FDA should not be promised or implied. Should describe any compensation or medical treatments that will be provided if injury occurs. Should also indicate whether the subjects will be billed for the cost of medical treatments.

130
Elements of Informed Consent
Explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights and whom to contact in the event of a research-related injury. Statement that participation is voluntary, that refusal to participate will involve no penalty or loss of benefits, that the subject may discontinue participation at any time without penalty. 3 components: a name of a specific office or person and the telephone number to contact for questions about 1) the research subjects’ rights 2) a research-related injury and 3) the research study itself. This may be a single person/phone number. The subjects must be informed that they may decline to participate or to discontinue participation at any time without penalty or loss of benefits.
 the research subjects’ rights 2) a research-related injury and 3) the research study itself. This may be a single person/phone number. The subjects must be informed that they may decline to participate or to discontinue participation at any time without penalty or loss of benefits.")
131
Elements of Informed Consent
The following elements should be included if appropriate: Statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant) which are currently unforeseeable. Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent. Depending on the amount of animal data available, these statements may need to be added, i.e. if the mutagenicity and teratogenicity studies have not yet been conducted/completed in animals. If measures to prevent pregnancy should be taken while in the study, that should also be included. They should be informed of circumstances for which their participation may be terminated without the subject’s consent. A statement that the investigator may withdraw subject if they do not “follow study procedures” is not appropriate. They may be informed that they will be withdrawn if they do not follow the instructions given to them by the investigator.
 which are currently unforeseeable. Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent. Depending on the amount of animal data available, these statements may need to be added, i.e. if the mutagenicity and teratogenicity studies have not yet been conducted/completed in animals. If measures to prevent pregnancy should be taken while in the study, that should also be included. They should be informed of circumstances for which their participation may be terminated without the subject’s consent. A statement that the investigator may withdraw subject if they do not follow study procedures is not appropriate. They may be informed that they will be withdrawn if they do not follow the instructions given to them by the investigator.")
132
Elements of Informed Consent
Any additional costs to the subject that my result from participation in the study. The consequences of a subjects’ decision to withdraw from the research and procedures for orderly termination of participation by the subject. 4. Explain any withdrawal procedures that are necessary for the subject’s safety and specifically state why they are important to the subject’s welfare.

133
Elements of Informed Consent
A statement that significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation will be provided to the subject. The approximate number of subjects involved in the study. 6. IF the numbers of subjects in a study may influence the subjects’ decision to participate.

134
The Consent Process The clinical investigator is responsible for ensuring that informed consent is obtained from each subject prior to their participating in the study. FDA does not require that the investigator actually obtain the informed consent. May delegate it to appropriate individual who is knowledgeable about the research. The investigator retains ultimate accountability.

135
The Consent Process The subject should date & sign the document.
Time of consent should be recorded. A copy of the consent form must be provided to the subject. A note is written in the progress notes confirming the consenting process. The signed consent form must be retained in the study records. The subjects dating the consent permits verification that the consent was actually obtained before they started the study. FDA does not require the subject’s copy to be a signed copy of the consent form, although this is preferred.

136
Informed Consent 2 types of forms
Written consent document that includes all the elements of informed consent required by 21 CFR Short form written consent stating that the required elements of informed consent have been presented orally. When the short form is used, there must be a witness to the oral presentation. The witness must be present for the entire discussion, not just the signing of the form. Also, the IRB must approve a written summary of what is to be said to the subject.

137
Informed Consent Exceptions
Both the investigator and a physician not participating in the investigation must certify in writing that the following conditions have been met. Subject is in a life threatening situation Consent cannot be obtained from the subject Insufficient time to obtain consent from the subject’s legal representative No alternative treatment that would have an equal or greater chance of saving the patient is available The test article is required to save the life of the subject.

138
Informed Consent Exceptions
Documentation of both the investigator and independent physician must be submitted to the IRB within 5 working days. Planned study that must be done in the emergency room in order to evaluate use of the test article in that setting. Limited to situations where intervention is life-saving and there is not time to obtain consent. Required FDA approval, IRB approval, and public disclosure.

139
Informed Consent Issues
Disclosing information to subjects requires common sense. Too much information can be as bad as too little. Too much can interfere with the subject’s ability to understand what is truly important. Need to provide enough information to make an educated decision.

140
Informed Consent Issues
Poor comprehension of consent documents is usually a result of low readability and excessive length. General population reads at a 6th or 7th grade level. Retention of information can be improved with repetition. Using a variety of methods such as individualized discussion and visual aids may increase retention and understanding. Improve readability by using short sentences and short words.

141
Informed Consent Issues
To demonstrate comprehension a subject must understand: Their condition The nature of the proposed treatment Alternatives to treatment Consequences of accepting or rejecting proposed treatment Risks and benefits of various options

142
Informed Consent Issues
Sufficient time must be given for the consent process. Sufficient time to read and digest the consent. Sufficient time to ask questions of staff. Sufficient time to consult with a friend or relative if appropriate. Sufficient time to reflect on his/her decision.

143
Informed Consent Issues
Informed consent must be obtained prior to participation in the study. Prior to any specific exams or screening procedures specific to the study to determine eligibility. Prior to discontinuing any existing medications the subject is taking to meet inclusion/exclusion criteria. (wash-out period) Procedures that are performed as part of the practice of medicine and which would be done regardless of the study, may be performed and the results used to determine eligibility.
 Procedures that are performed as part of the practice of medicine and which would be done regardless of the study, may be performed and the results used to determine eligibility.")
144
Study Payment Payment for subjects is viewed as a recruitment incentive not a benefit. Financial incentives are generally used when benefits to the subject are remote or non-existent. Amount and schedule of payment should be presented to the IRB at the time on initial review. IRB should review the amount of payment and timing of payment to assure that they are not coercive or do not present undue influence.

145
Study Payment Payment should accrue as the study progresses.
Payment may not be contingent upon the subject completing the entire study. Payment to subjects who withdraw may be made at the time they would have completed the study (or completed a phase of the study). Payment of a small proportion as an incentive for completion of the study is acceptable as long as it is not coercive. Payment details must be included in the informed consent.
. Payment of a small proportion as an incentive for completion of the study is acceptable as long as it is not coercive. Payment details must be included in the informed consent.")
146
Informed consent must be approached as a process rather than a distinct event in time.

147
Assess Competence to Consent
Ability to Express a choice Ability to Understand information about a trial Ability to Reason with relevant information to understand a logical process of weighing options

148
What Can We Do? When obtaining consent ask yourself…..
“Is the subject adequately informed to understand and comprehend the information in order to make a conscious decision to participate in a clinical research trial?”

149
What Can We Do? Reduce boilerplate language
Extended discussion about the trial Multimedial presentations Enhanced forms “Test” of knowledge

150
The Ability to Understand the Informed Consent
It is a function of: Intelligence Rationality Maturity Language Ethically it is the investigator’s responsibility to ensure that the research subject has comprehended the information.

151
What Can We Do? Use less text and more graphics.
Simplify language – use action verbs and direct statements Read the FDA information sheets Substitute long wordy sentences with tables or lists. Develop a list of lay terms Make the form “EYE” friendly

152
Sample Phrases All blood samples will be drawn from a forearm vein via an intravenous catheter contra lateral to the one used for drug administration in the case of the continuous infusion regimens. We will need to take some blood from your arm for this study. We will insert a needle attached to a plastic tube in your forearm. A second needle with a plastic tube will be placed in your opposite arm

153
What Can We Do? How the document is laid out is just as important to readability and comprehension as the information it contains Use a written or verbal post-consent evaluation to determine if the critical points were understood (must be IRB approved)
")
154
Informed Consent Questions??
Is getting the subject to sign a consent document all that is required by the regulations?? No!! The consent document is just a written summary of the information that should be provided to the subject. The consent form is just a guide to use for the discussion. The process should give the subject adequate information about the study, provide adequate opportunity for the subject to consider all their options, respond to the subject’s questions, ensure that the subject has understood all the information, obtain the subject’s voluntary agreement to participate, and continue to provide information as the subject or situation requires.

155
Informed Consent Questions??
May informed consent be obtained by telephone from a legal representative? No. This does not satisfy the regulations. A document can be faxed to the representative. The discussion can be conducted by telephone and then the signed form can be faxed back.

156
Informed Consent Questions??
Does the copy of the consent form given to the subject have to be a signed copy? No! This is encouraged but not mandatory. The purpose of this is to allow the subject to review the information with others as well as to serve as a continuing reference for them.

157
Informed Consent Questions??
Who should be present when the informed consent interview is conducted. FDA does not require a third person or witness. The person who conducts the consent interview should be knowledgeable about the study and able to answer questions. Investigator may delegate responsibility to a person who has received appropriate training to perform this activity. Some sponsors and some IRBs require the investigator to personally conduct the interview. R

158
Informed Consent Questions??
When should study subjects be informed of changes in the study? Those subjects who are enrolled and actively participating in the study should be informed of any changes if it may affect to their willingness to continue their participation in the study. FDA does not require reconsenting subjects that have completed their active participation or who are still actively participating but the change will not affect their participation. If a change will not affect current subjects, e.g. a dose change for future participants or a change in procedures for new subjects, they do not need to be reconsented.

159
IRBs

160
Sound Clinical Research Depends on Compliance with …
Good Clinical Science (Industry and Investigator) + Good Statistical Design (Industry and IRB) Sound Ethical Conduct (IRB, Investigator, Industry)
 + Good Statistical Design. (Industry and IRB) Sound Ethical Conduct. (IRB, Investigator, Industry)")
161
What is an IRB? Knowledge and application of commitments, regulations, guidelines, state and local laws and standards of professional conduct Knowledgeable about the care and protection of vulnerable population An appreciation for doing the right thing

162
What is an IRB? Under FDA Regulations (21 CFR 56 - Subpart C) and (45 CFR 46 - Subpart A) An IRB is an appropriately constituted group that has been formally designated to review and monitor biomedical and behavioral research involving human subjects.

163
What is an IRB? An IRB has the authority to approve, require modifications (in order to secure approval), or disapprove search. An IRB is responsible for ensuring, in advance and by periodic review, that steps are taken to adequately protect the rights and welfare of human subjects.

164
Why an IRB? Knowledge and application of commitments, regulations, guidelines, state and local laws and standards of professional conduct Knowledgeable about the care and protection of vulnerable population An appreciation for doing the right thing.

165
Institutional Review Boards
Membership At least five members with varying backgrounds. Must consist of both men and women. May not be entirely of members of one profession. At least one member whose primary concerns are in a scientific area and one member in a non-scientific area. At least one member who is not otherwise affiliated with the institution. No IRB may have a member participate in the IRBS review of any project in which they have a conflicting interest. May bring in an expert to assist in review of complex issues but they may not vote. Members may satisfy more than one category requirement.

166
Institutional Review Boards
IRBs must: Follow written procedures Review research at convened meetings at which a majority of the members (quorum) is present including at least one member whose primary concerns are in nonscientific areas.
 is present including at least one member whose primary concerns are in nonscientific areas.")
167
What is the Role of the IRB?
Protect the rights and welfare of human research subjects. Answer three basic questions: Should the study be done at all? Do the benefits outweigh the probable risks and is this information adequately conveyed in the consent form? How will the research subject be protected on an ongoing basis?

168
Institutional Review Boards
Data submitted to IRB for review: Protocol, protocol amendments Informed consent form Advertising and recruitment process Information on the drug e.g. Investigator’s brochure Information on the investigator e.g. CV Should review any written information that will be provided to the subjects. Should also review the methods and material that investigators propose to use to recruit subjects.

169
What is the Role of the IRB? Criteria for IRB Approval
Documentation of informed consent Monitoring of data Privacy and confidentiality Additional safeguards of all vulnerable populations Risks v Benefits Risks minimized Equitable selection Informed consent Investigator training relative to responsibilities

170
IRB Decisions Authority to make three possible decisions regarding studies: Approved Disapproved Requires modifications to be approved Notification of decisions must be made in writing to investigator. Continuing review of research must be done at a minimum of once per year. The interval for continuing review can be less than once per year if appropriate to the degree of risk.

171
The IRB “A collegial and cooperative relationship”
Judge Protector Facilitator Enforcer Educator Colleague Partner

172
Institutional Review Boards
The purpose of IRB review is to assure that appropriate steps are taken to protect the rights and welfare of humans participating as subjects in the research. Risks to subjects are minimized Risks to subjects are reasonable in relation to anticipated benefits Selection of subjects is equitable Informed consent will be sought Research plan makes adequate provisions for monitoring the data collected to ensure the safety of subjects Adequate provision to protect the privacy of subjects Ask the questions Should the study be done at all? Do the benefits outweigh the probable risks and is this information adequately conveyed in the consent form? How will the research subject be protected on an ongoing basis?

173
Focusing on Continuing Review: Why?
Has any significant and/or new information been made available since the study received last approval? Do the research subjects need different information with which to reconsider their continued participation Is the study being conducted as approved? Did the IRB use the same review and approval process that it used to conduct the initial review?

174
Information required by the IRB for Continuing Review
Enrollment and study start and stop Adverse event information and follow-up where indicated Study progress reports Review of consent process and document changes. Any new findings important to the subject’s safety Unanticipated problems that pose a risk to the subject

175
Institutional Review Boards
Expedited review: Procedure to review and approve research without convening a meeting of the IRB. Used for minor changes in previously approved research. Review is done by the IRB chairperson or by one of the experienced members of the IRB May not be disapproved by expedited review—only be full review.

176
HIPAA Requirements

177
What is HIPAA Health Insurance Portability and Accountability Act
Signed August, 1996 Congress mandated that the Secretary of HHS adopt standards to facilitate the electronic exchange of health information

178
Who Must Comply with HIPAA?
Health Plans, health care clearinghouses, and health care providers that transmit health information electronically in connection with defined HIPAA statute Transmissions related to … Health care claims Health care payment or remittance advice Coordination of benefits Health clam status Enrollment and disenrollment in a health plan, Eligibility for health plan Health plan premium payments Referral certification and authorization First report of injury Health claims attachments Other transactions

179
What is a Covered Entity
Employees of a covered entity (hospital, etc), then they must comply with regulation. Function independent of a covered entity (i.e. physician who is not employee of the hospital), they must meet all of the requirements independent of the institution.
, then they must comply with regulation. Function independent of a covered entity (i.e. physician who is not employee of the hospital), they must meet all of the requirements independent of the institution.")
180
What about Researchers
Research is not a covered function, however…. If the research involves either the provision of health care by a covered entity Retention of medical records or biological samples by a covered entity ….then disclosure of information or clinical trial data for research purposes must comply with the Privacy Rule

181
Penalty for Non-Compliance
Civil monetary penalty of $100 per occurrence ($25K max) Federal Criminal penalties for persons who knowingly obtain or disclose health information in violation of Privacy Rule.
 Federal Criminal penalties for persons who knowingly obtain or disclose health information in violation of Privacy Rule.")
182
What is Protected Health Information?
Either oral or recorded information in any form or medium Is created or received by a health care provider, health plan, etc. Provision of health care to an individual

183
What is De-Identification?
De-Identification information is no longer covered by the HIPAA requirements. All identifying information has been removed including Names Geographic subdivisions smaller than a state (i.e., no city, no zip code), except for the initial three digits of the zip code if, according to the current publicly available data from the Bureau of the Census, the geographic unit contains more than 20,000 people Any date (except year; i.e., no month or day of month) For subjects older than 89 years of age, specific age may not be mentioned Telephone number Fax number address
, except for the initial three digits of the zip code if, according to the current publicly available data from the Bureau of the Census, the geographic unit contains more than 20,000 people. Any date (except year; i.e., no month or day of month) For subjects older than 89 years of age, specific age may not be mentioned. Telephone number. Fax number. address.")
184
What is De-Identification?
Social security number Medical record number Health plan beneficiary number Any other account numbers Certificate or license numbers Vehicle identification number Medical device identification or serial number Personal website URL Internet protocol (IP) address Fingerprint, voiceprint, or other biometric identifiers Full-face photographic images Any other unique identifying number, characteristic, or code
 address. Fingerprint, voiceprint, or other biometric identifiers. Full-face photographic images. Any other unique identifying number, characteristic, or code.")
185
Impact of Reviewing Records for Research Participation
Allows covered entities to permit researchers to review PHI held in medial records or elsewhere for “review Preparatory to research” for the purpose of assessing the pool of potentially eligible subjects. Contact of these subjects is related to whether the individual is a member of the covered entities workforce

186
Other Issues Adverse events may be reported to research sponsors, public health agencies and health oversight agencies without obtaining authorization IRBs, DSMBs, or other investigators should be identified in the original authorization form.

187
Authorization Core Elements
A description of the PHI to be used or disclosed, The names or other specific identification of the person or persons (or class of persons) authorized to make the requested use or disclosure The names or other specific identification of the person or persons (or class of persons) to whom the covered entity may make the requested use or disclosure A description of each purpose of the requested use or disclosure Authorization expiration date or expiration event that relates to the individual or to the purpose of the use or disclosure ("end of the research study" or "none" are permissible for research, including for the creation and maintenance of a research database or repository) Signature of the individual and date.
 authorized to make the requested use or disclosure. The names or other specific identification of the person or persons (or class of persons) to whom the covered entity may make the requested use or disclosure. A description of each purpose of the requested use or disclosure. Authorization expiration date or expiration event that relates to the individual or to the purpose of the use or disclosure ( end of the research study or none are permissible for research, including for the creation and maintenance of a research database or repository) Signature of the individual and date.")
188
What does this mean practically speaking?
In the clinic use first names only Sign-in sheets should have cover over previous names Limit access to the clinic to essential personnel only Name tags should only have first name Screen saver on clinic computers, if first and last names are displayed

189
In Summary Historical misuse and abuse Key Investigator responsibility
Informed consent is critical component IRB oversight Patient Privacy

190
Questions!!!!!!

191
Drug Development Process
care Drug Development Process community Donna W. Dorozinsky, RN, MSN, CCRC research teach

192
Course Objectives Discuss the differences in Phase I, II and III of the drug development process. Identify the key requirements of a New Drug Application. Discuss the different types of studies done at the CPU

193
Introduction: Only 5 in 5,000 compounds that go into pre-clinical make it to human testing. It takes years from conception of a new drug to its approval Time is money It takes 10 to 12 years from conception of a new drug to its approval. By this time, it has only a relatively short time left on its patent. Time is money. To help reduce these timelines, the FDA has been working with pharmaceutical companies to “fast track” certain drugs

194
$1B/year in sales $2.7m/day 2 week delay = $38m

195
Drug Development Costs
Triangle Pharmaceuticals Cumulative Drug Development Expenses from This graph represents Cumulative drug development expenses for Triangs Pharmaceuticals. This company in-licenses compounds and focuses on developing these compounds. Costs represent clinical study conduct only. It has been estimated that one drug can cost as much as $800 million to develop. That does not include all of the costs in developing drugs that are never approved. Shows how drug development costs continue to skyrocket each year due to drug synthesis and manufacturing costs, clinical trial costs, employee compensation, and pre-clinical testing. Drug development costs are now over $250 million dollars.

196
Drug Development Costs
2003 as reported by University $897M 2006 as reported by CEO at Lilly $1.2B 2010 forecast by CEO at Lilly $2B

197
Contributions to Rising Costs
Soaring R&D costs Lowered drug approvals Increased development times The loss of patent protection on several blockbuster Safety issues Pricing pressures.

198
Drug development difficulties:
Drugs usually do not cure diseases. Diseases don’t follow a predictable path Sometimes measurements of a disease are subjective Drugs usually do not cure diseases. They only reduce the risk of death but don’t eliminate it (e.g. AIDS drugs) or relieve the symptoms of the illness (e.g. pain) or alter a clinical measurement (e.g. blood pressure, blood glucose levels) Measurements are subjective in situations of pain. Much research is being done in area of pain management. Pain is a subjective symptom.
 or relieve the symptoms of the illness (e.g. pain) or alter a clinical measurement (e.g. blood pressure, blood glucose levels) Measurements are subjective in situations of pain. Much research is being done in area of pain management. Pain is a subjective symptom.")
199
Drug Development Steps
Pre-clinical (animal) testing Filing Investigational New Drug Application (IND) with the FDA Phase 1 studies Phase 2 studies Phase 3 studies New Drug Application (NDA) Drug approval by FDA It is common to have an overlap of phase I into Phase II and III. Submission of a New Drug Application (NDA) FDA reports whether drug is approved or not
 testing. Filing Investigational New Drug Application (IND) with the FDA. Phase 1 studies. Phase 2 studies. Phase 3 studies. New Drug Application (NDA) Drug approval by FDA. It is common to have an overlap of phase I into Phase II and III. Submission of a New Drug Application (NDA) FDA reports whether drug is approved or not.")
200
Drug Development Steps
Pre-clinical (animal) testing Filing Investigational New Drug Application (IND) with the FDA Phase I studies Phase II studies Phase III studies New Drug Application (NDA) Drug approval by FDA It is common to have an overlap of phase I into Phase II and III. Submission of a New Drug Application (NDA) FDA reports whether drug is approved or not
 testing. Filing Investigational New Drug Application (IND) with the FDA. Phase I studies. Phase II studies. Phase III studies. New Drug Application (NDA) Drug approval by FDA. It is common to have an overlap of phase I into Phase II and III. Submission of a New Drug Application (NDA) FDA reports whether drug is approved or not.")
201
© ***Note overlap of phases of development
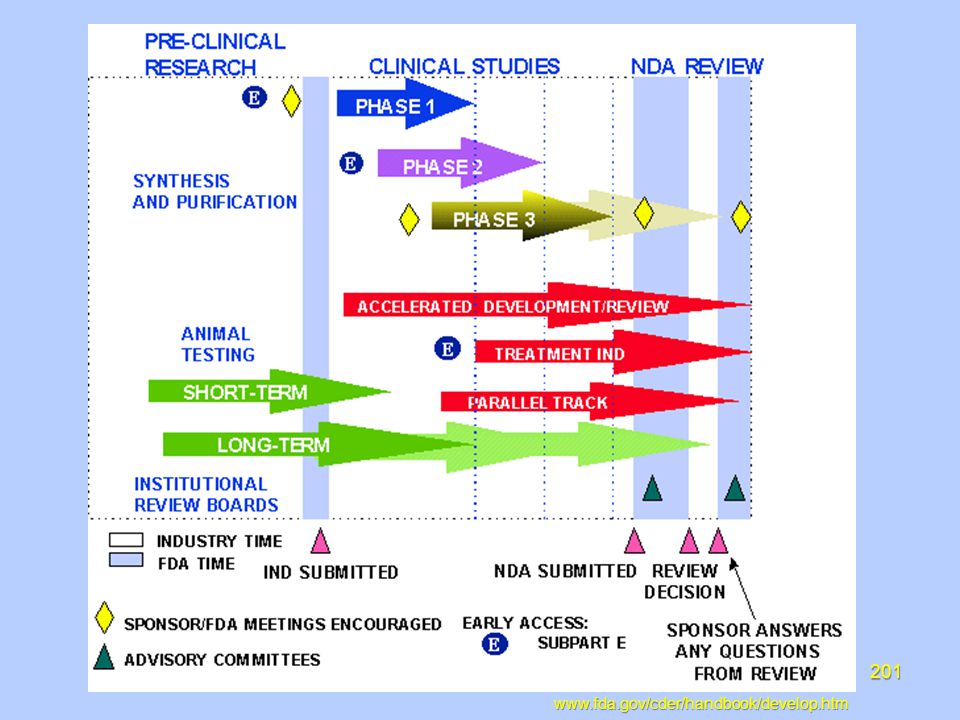
202
Pre-Clinical Testing Pre-clinical studies are studies that test a drug on animals and other non-human test systems Start with initial chemical development of a compound The pre-clinical testing of a compound can take years. Animal testing in at least 2 species must be done before progressing to the human phase. There needs to be a good understanding of the PK profile of the compound in animals. While animal data does not always predict human response, it can be a good indicator.

203
Pre-Clinical Testing Studies are done with cell tissues, isolated tissues, and animals to determine: Pharmacologic effects Toxic effects LD-50 (lethal dose) ADME Toxic effects including mutagenicity—The capacity of a chemical or physical agent to cause permanent genetic alterations teratogenicity-- The capacity of a chemical or physical agent to cause birth defects or abnormalities. Carcinogenicity—The capacity of a chemical or physical agent to cause cancer. LD50-(lethal dose)—the dose which kills 50% of the animals
 ADME. Toxic effects including. mutagenicity—The capacity of a chemical or physical agent to cause permanent genetic alterations. teratogenicity-- The capacity of a chemical or physical agent to cause birth defects or abnormalities. Carcinogenicity—The capacity of a chemical or physical agent to cause cancer. LD50-(lethal dose)—the dose which kills 50% of the animals. ©")
204
Pre-Clinical Testing ADME
Absorption: how it goes from being a pill or liquid to a biologically available form. (a form the body can actually use) Distribution: getting the drug to the tissue Metabolism: (biotransformation) getting the drug from the biologically available form to a more water soluble form Excretion: moving metabolites from tissue to circulation to organs of excretion Kidneys Liver bowel Absorption: Uptake from site of administration to systemic circulation Route of administration—oral, sub-cutaneous, intramuscular, intravenous, topical, etc Distribution: Distribution of drug from blood to site of action Metabolism: Sum of chemical reactions transforming substances Excretion: Final elimination from tissues and systemic circulation of the body, From kidney into urine; from liver into bile and feces Other routes: airway mucus, skin sweat, hair, milk
 Distribution: getting the drug to the tissue. Metabolism: (biotransformation) getting the drug from the biologically available form to a more water soluble form. Excretion: moving metabolites from tissue to circulation to organs of excretion. Kidneys. Liver. bowel. Absorption: Uptake from site of administration to systemic circulation. Route of administration—oral, sub-cutaneous, intramuscular, intravenous, topical, etc. Distribution: Distribution of drug from blood to site of action. Metabolism: Sum of chemical reactions transforming substances. Excretion: Final elimination from tissues and systemic circulation of the body, From kidney into urine; from liver into bile and feces. Other routes: airway mucus, skin sweat, hair, milk.")
205
Pre-Clinical Testing Must think about marketing considerations before going to Phase I What is the intended population and what is the incidence in that population? Do competitors have similar compounds in their pipeline? What are current therapy options for this disease and are they effective and safe? Are there any unique or disqualifying toxicology results for this specific compound? What will be the likely route of administration? What will be the dosing frequency? In today’s competitive environment and with the issues of reimbursement, there is not room for a “me too” Once day dosing via oral route is most viable option. For diseases like decreased bone density, even less frequent dosing is the norm.

206
The IND When… Filed prior to human dosing
If no response from the FDA, the clinical trial may start 30 days after it is received. Annual IND update.

207
The IND Why…. FDA to ensure the safety and rights of subjects
In Phase 2 and 3, help assure that the quality of scientific evaluation is adequate to evaluate safety and efficacy. Allow for shipping of drugs across state lines.

208
Investigational New Drug Application (IND)
An IND is Submitted to the FDA demonstrating there is reasonable justification for studying the drug in humans Includes results of pre-clinical testing Shows plan for human testing Must be obtained before the first dose in humans Must include the results of pre-clinical testing in animals Must show the development plan for human testing

209
Investigational New Drug Application (IND)
Includes Table of contents Introductory statement General investigational plan Investigators Brochure Protocols including study protocols, investigator data, facilities data, IRB data Chemistry, manufacturing and controls (CMC) Pharmacology and Toxicology information Previous human experience, if any Additional information including drug dependence and abuse potential Any other relevant data These are the parts to the actual IND application.
 Pharmacology and Toxicology information. Previous human experience, if any. Additional information including drug dependence and abuse potential. Any other relevant data. These are the parts to the actual IND application.")
210
Investigational New Drug Application (IND)
Becomes effective 30 days after it is received by the FDA Updated annually and amended to reflect protocol changes, additional protocols, and information about serious adverse events FDA decides if it is safe to move on to testing in humans Generally at BMS you dose on day 31 or 32. However, the FDA can come to you on Day 30 and put the compound on hold. At that point you can not dose in humans.

211
Clinical Trials Human studies designed to distinguish a drug’s effect from other influences. Necessary to determine if a drug is safe and effective, establish dose efficacy, and identify side effects Best way science has come up with to determine what a new drug really does It is important to test drugs in the population of people that they are meant to help Need to design clinical studies that ask and answer the right questions Defined as human studies designed to distinguish a drug’s effect from other influences—for example, a spontaneous change in disease progress or the effect of a placebo.

212
Clinical Trials # of Patients Length Purpose
% of Drugs Successfully Tested Phase 1 20-100 Several months Mainly safety 70% Phase 2 Up to several hundred Several months to 2 years Some short-term safety but mainly efficacy 33% Phase 3 Several hundred to several thousand 1-4 years Safety, efficacy, dosage 25-30% For example, of 100 drugs for which investigational new drug applications are submitted To FDA, about 70 percent will successfully complete phase 1 and go on to phase 2; about 33 percent of the original 100 will complete phase 2 and go to phase 3; and 25 to 30 of the original 100 will clear phase 3 (and, on average, about 20 of the original 100 will ultimately be approved for marketing).
.")
213
Study Design Terminology
Blinding Unblinded or open-label Single Blind Double-blind Unblinded or Open-Label--Subject, investigator, and evaluator know the treatment Single Blind—The subject does not know the treatment Double-blind—The subject and the investigator do not know the treatment Double-Dummy—Used to bind two agents dissimilar in appearance; subjects take both agents

214
Study Design Terminology
Subject Assignment Randomization – assigned by chance Stratification – assigning someone to a treatment group based on their sex, weight, age, disease state Cross-over design – Each patient receives both treatments Parallel Design – Patients are randomized to one of two treatment groups and usually receive the same treatment through-out the entire study Randomization—method used to equitably assign study subjects to treatment to eliminate bias Stratification—method used to control assignment of subjects to treatment based on a variable which is thought to affect he study outcome

215
Phase I Primarily intended to determine the tolerability of the drug
Conducted in healthy volunteers Establishes a dosing range Establishes the route of administration Determines the most frequent adverse events Primarily intended to determine the tolerability and in particular, the highest dose with acceptable tolerability. There is tremendous risk to the drug approval if you choose the wrong dose for Phase III studies. You risk non-approval because of lack of efficacy or if you see toxicity you risk the future of the drug.

216
Phase I Determination of pharmacokinetics and pharmacodynamics
Pharmacokinetics (PK) What the body does to the drug ADME Usually measured in blood or urine Pharmacodynamics (P-dyne) What the drug does to the body E.g. heart rate, ECG, blood pressure Pharmacokinetics: the time course of a drug in a biological system Pharmacodynamics: Physical effects of a drug What the drug does to the body e.g. heart rate, blood pressure, ECG
 What the body does to the drug. ADME. Usually measured in blood or urine. Pharmacodynamics (P-dyne) What the drug does to the body. E.g. heart rate, ECG, blood pressure. Pharmacokinetics: the time course of a drug in a biological system. Pharmacodynamics: Physical effects of a drug. What the drug does to the body. e.g. heart rate, blood pressure, ECG.")
217
ADME

220
Phase I Usually start with single dose study
Dose escalation to maximum tolerated dose Multiple ascending repeat dose studies Kinetic profile is established Drug interactions QTC intervals Looks at no effect level, efficacious or desired level, toxic level For oncology and HIV drugs, usually use patients instead of healthy subjects. Use patients because of drug toxicities. Also a push to get into patients early in phase I to look for a signal of efficacy. For example, in diabetes do an early repeat dose rising looking at 2-3 doses in a small sample size. May look at serum markers that suggest better glucose management. Companies are conducting single ascending dose study, multiple repeat ascending dose study and Proof of Concept then moving to phase II. This is a trend in an effort to reduce drug development time and reduce progression of drugs that do not have a future. QTC studies are now routinely part of Phase 1 studies as per new FDA guidelines.

221
Phase I Future is in establishing early signaling of drug efficacy.
Proof of Concept often conducted in Phase I then move to phase II while completing drug interactions, food interactions, etc.

222
Phase II Primary objective
To examine safety and therapeutic effectiveness in patients Proof of concept--To find out whether the drug works in people who have the targeted disease or condition.

223
Phase II Goals Determine dose and regimen
Evaluate potential study endpoints Evaluate therapeutic regimens including concomitant medications Evaluate target population including disease severity Determine all of above for further study in phase 2 or 3

224
Phase II May be divided into two phases
Phase 2A: proof of concept pilot studies Phase 2B: Demonstrate efficacy Assess short term safety Dose finding/dose ranging Uses a few dozen to 300 subjects Duration: short to medium length lasting up to a few months This is where you are now seeing a blending of phase I and phase II. Phase 2A is moving back into phase I. With the push to decrease time to develop new drugs, more studies, including proof of concept and efficacy, are being moved into phase 1.

225
Phase II Information defined in Phase 2 Safety in patients Efficacy
Pharmacodynamics Pharmacokinetics Bioavailability Drug/disease interactions Drug/drug interactions Efficacy at different doses Pediatric information The information in Phase 2 is in addition to the data in phase 1. e.g. Safety, PK, PD, bioavailability, is also done in phase 1. Information defined in Phase 2 builds on the data already defined in Phase 1. Some data is new, while some is merely a continuation of the data found in phase 1. Recent push in the FDA to evaluate safety and efficacy in the pediatric population.

226
Phase III Primary objective
To confirm therapeutic effectiveness and safety in a less restricted patient group Consists of well-controlled trials to support marketing approval by confirming preliminary data collected in phase 2 on safety and efficacy in intended patient population

227
Phase III Uses several hundred to 3000 subjects Duration:
parallels anticipated treatment May be several years for chronic conditions Takes place at multiple centers including hospitals and doctors offices Study design has broader patient eligibility and may have 2-3 treatment groups Tests final formulation of drug

228
Phase III Information typically defined:
Efficacy and safety in population subgroups including different disease stages Dosing interval Dose response relationship Efficacy with another drug (drug combination therapy) Drug/drug interactions Drug/disease interactions Risk/benefit information
 Drug/drug interactions. Drug/disease interactions. Risk/benefit information.")
229
Pravachol Approval Pravachol Primary Prevention Study, also known as the West of Scotland study, was pivotal phase III study used to file NDA The Pravachol Primary Prevention Study, evaluated the use of Pravachol in 6,595 subjects over a five-year period, demonstrated that Pravachol reduced the risk of first heart attack by 31%. Death from cardiovascular disease was reduced by 32%, and there was no increase in death from non-cardiovascular causes. The study showed that Pravachol reduced the need for coronary procedures such as balloon angioplasty and bypass surgery by 37%. Another major finding from the Pravachol Primary Prevention Study was that Pravachol benefit, in terms of reduction in first heart attacks, begins at about six months after the initiation of therapy. This is information on Pravachol, a drug that was developed out of this Unit. Look at this information and think about what data was key in getting the drug aproved.

230
The NDA What…. Formal step a drug sponsor takes to receive FDA approval to market a new drug in the USA Application to market a new drug under Section 505 of FDA. Establishes an efficient and thorough drug review process

231
When? Once all pre-clinical and clinical reports are available to support the file.

232
New Drug Application (NDA)
Includes: Index Overall summary of drug and study results Technical sections Chemistry, manufacturing, and controls (CMC) Non-clinical pharmacology and toxicology Human PK and availability Clinical microbiology if applicable Clinical data Statistics
 Non-clinical pharmacology and toxicology. Human PK and availability. Clinical microbiology if applicable. Clinical data. Statistics.")
233
New Drug Application (NDA)
Includes Case Report Tabulations Case Report Forms Patient information and certification Establishment description Environmental assessment or exemption Includes samples of the drug Typically consists of over 100,000 pages and may contain data on more than 3000 patients

234
New Drug Application (NDA)
Once the NDA is filed, the FDA has 60 days to decide whether to file it for review FDA’s Center for Drug Evaluation and Research (CDER) will then review the drug for approval May be refused if data is viewed as incomplete by the FDA
 will then review the drug for approval. May be refused if data is viewed as incomplete by the FDA.")
235
New Drug Application (NDA)
Review team consists of: Chemists Pharmacologists Physicians Pharmacokineticists Statisticians Microbiologists Review team consists of Chemists: focus on how the drug is made, the manufacturing controls, and the packaging. Make sure they are adequate to provide quality and purity of the product Pharmacologists: evaluate the effects of the drug on animals in short-term and long-term studies Physicians: Evaluate the results of the clinical tests including the drugs adverse effects as well as the therapeutic effects. Evaluate whether the proposed labeling is accurate as to the effects of the drug Pharmacokineticists: Evaluate the ADME of the drug Statisticians: Evaluate the designs for each study, the analyses and conclusions of safety and effectiveness based on the study data Microbiologists: Evaluate the data on anti-infectives.

236
New Drug Application (NDA)
Team evaluates the drug to see if it is “safe” and effective for proposed use “Safe” means the benefits of the drug outweigh the risks They analyze the results and look for any weaknesses of study design or analyses They determine if they agree with the sponsors or need additional information

237
New Drug Application (NDA)
Prescription Drug User Fee Act passed in 1992 to provide funding in an effort to reduce FDA approval time and bring drugs to market faster. CDER’s goal is to review and act on at least 90% of NDAs for standard drugs no later than 10 months after the NDA is received and no later than 6 months for priority drugs Standard drug: Drugs that offer only a minor improvement or no improvement over drugs currently on the market Priority drug: a drug believed to represent potential major advances in healthcare Priority given to May a new dosage form of or a new use for an existing drug.

238
New Drug Application (NDA)
FDA has worked to shorten time devoted to clinical trials by Having meetings with drug companies to streamline studies and eliminate duplicate studies Expediting approval of innovative agents As part of the review process, on-site inspections are made of some of the clinical sites As part of the review process, on-site inspections are made of some of the clinical sites to verify that the data/work cited in the NDA is valid

239
New Drug Approval Time by Year Median of Total Approval Times (in months)
The amount of time that it takes to get approval has been decreasing over time.

241
New Drug Application (NDA)
Advisory committee Panel of outside experts Review the safety and efficacy results CDER is does not have to take the committees recommendations but they usually do Sometimes the FDA puts together an advisory committee to review the safety and efficacy results. This is a panel of outside experts. CDER is not bound to take the committee recommendations but it generally does.

242
New Drug Application (NDA)
Three possible results Approved Approvable: drug can probably be approved after resolution of certain issues Not approvable Approvable: drug can probably be approved after resolution of certain issues. May be labeling issues, manufacturing issues or a need for additional data

243
IND & NDA As you look at this cartoon you see the many different perspectives of the glass in the center of the page. Each of these perspectives add a dimension to the development of the compound.

244
New Drug Approval Time by Year Median of Total Approval Times (in months)
The amount of time that it takes to get approval has been decreasing over time.

245
Label When the FDA approves a drug, it approves the label for the drug
The label is what appears on the package insert for the drug. This information also appears in the PDR. Many of the studies done in Phase I contribute to the content of the label or package insert.

246
Label Label includes: Description of the drug Clinical Pharmacology
Chemical class and structure Pharmaceutical action Product presentation Clinical Pharmacology Mechanism of action Pharmacodynamics Pharmacokinetics and metabolism PK data is key piece of Phase I program. This is an important part of the labeling information.

247
Label Label includes Indications and usage Contraindications
Warnings, WARNINGS Precautions Drug interactions Carcinogenesis Mutagenesis Fertility Effect Nursing Mothers Pregnancy Children Elderly The CPU does many of the drug interaction studies that provide data for the Precautions.

248
Label Label includes Adverse Reactions
By organ or body system Stratification by % incidence Drug abuse and dependency potential Overdosage Dosage and Administration How supplies Adverse Events from Phase I are captured as Adverse Reactions on the label.

249
Label May include a Black Box warning
This is a separate warning that is highlighted for the drug May be mandated by the FDA as terms of approval Not desirable In order to achieve approval the company must agree to place this black box warning on the label of the drug. Appears on the package insert in large black box.

250
Phase IV These are studies that occur after a drug has been approved
May explore long term effects May explore how participants respond to different dosages May compare with competitors products to support marketing claims May look at the incidence of adverse reactions These are often done at the request of the FDA.

251
Questions ??????

252
Legal Issues in Research
care Legal Issues in Research community Priya Sankar, Esq. Assistant General Counsel Associate Director, Contracts and Budgets research teach

253
Red Flag

254
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Other Legal Issues 254

255
Type of Clinical Research
Internal Industry Sponsored Government-Funded (e.g. NIH) 255
 255.")
256
Public Health Service (PHS )
Department of Health & Human Services (HHS) National Institute of Health (NIH) Food and Drug Administration (FDA) Center for Medicare & Medicaid Services (CMS) Center for Disease Control (CDC) Agency for Healthcare Research & Quality (AHRQ) Agency for Toxic Substances and Disease Registry (ATSDR) Health Resources & Services Administration (HRSA) Substance Abuse and Mental Health Services (SAMHSA) 256
 National Institute of Health. (NIH) Food and Drug Administration. (FDA) Center for Medicare & Medicaid Services. (CMS) Center for Disease Control. (CDC) Agency for Healthcare Research & Quality. (AHRQ) Agency for Toxic Substances and Disease Registry. (ATSDR) Health Resources & Services Administration. (HRSA) Substance Abuse and Mental Health Services. (SAMHSA) 256.")
257
What do they do? HHS Department of Health & Human Services
Protecting the health of all Americans and providing essential human services PHS Primary division of HHS Component of HHS that makes up the key agencies (e.g. NIH, FDA, etc.) OHRP Office of Human Research Protections Oversees national system of protecting human subjects in research (i.e. Institutional Review Boards) through regulations, oversight, guidance 257
 OHRP. Office of Human Research Protections. Oversees national system of protecting human subjects in research (i.e. Institutional Review Boards) through regulations, oversight, guidance")
258
What do they do? NIH NIH is the government’s research organization.
It supports 38,000 research projects nationwide in diseases including cancer, Alzheimer's, diabetes, arthritis, heart ailments and AIDS. Includes 27 separate health institutes and centers. FDA FDA assures the safety of foods and cosmetics, and the safety and efficacy of pharmaceuticals, biological products, and medical devices OIG Office of Inspector General Protect the integrity of Department of Health and Human Services (HHS) programs, as well as the health and welfare of the beneficiaries of those programs 258
 programs, as well as the health and welfare of the beneficiaries of those programs")
259
Key Players in Clinical Research
Study Subject St. Peter’s University Hospital Researcher PHS (HHS OHRP) NIH FDA Industry Sponsor IRB 259
 NIH. FDA. Industry Sponsor. IRB")
260
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Protection of Human Subject (HIPAA) 260
 260.")
261
Why Have A Contract? Anti-Kickback Safe Harbor Ensure Compliance
Protect Existing Rights Intellectual Property Confidential Information Allocate Rights and Obligation Publication Rights Intellectual Property (Inventions) Liabilities (Indemnification & Insurance) Cost 261261 261
 Liabilities (Indemnification & Insurance) Cost")
262
Confidentiality Agreements The Contract before the Research Contract
What is it? Usually required by Industry Synonyms: CDAs NDA Nondisclosure Agreements Secrecy Agreements 262

263
Key Terms Confidentiality Agreements
Defines what is “Confidential Information” Study Drug/Device Study Never undefined Defines what is not “Confidential Information: Public Information Already Know Already Given by Third Party Required to Disclose by Law Independently Developed 263

264
Key Terms Confidentiality Agreements
3. Defines how it will be disclosed 4. Defines who it may be disclosed to 5. Defines how long it shall be held confidential 264

265
Confidentiality Agreements
Protect Yourself! Protect your Colleagues! Know your institution’s policy Be careful what you sign Be careful what you open Make sure you are aware of your obligations. When in doubt, seek legal counsel You may be personally liable! 265

266
Clinical Trial Agreements
5 Key Provisions Intellectual Property Confidential Information Publication Indemnification Patient Injury Compensation (if clinical) 266
 266.")
267
Government Funded (e.g. PHS Funded Research)
Grants Funds transferred to awardee Work independently Report results at end Cooperative Agreements Substantial involvement Gov’t involved in planning and implementation Subcontracts to Grants/Cooperative Agreements

268
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Protection of Human Subject (HIPAA) 268
 268.")
269
What is Conflict of Interest?
Conflict of interest exists if it actually or potentially may have influence over the outcome of the research 269

270
Laws & Regulations Each agency has developed differing definitions of conflict of interest and how and to whom conflict disclosures should be made. 270

271
Laws & Regulations Neither the PHS or FDA regulations: 1. Identify specific conflicts of interest or financial interests that are prohibited. 2. Define the role of the IRB in dealing with conflict of interest. 271

272
PHS FUNDED RESEARCH 272

273
PHS Regulations (42 CFR § 50 ) (45 CFR § 94)
Applies to: PHS grants or cooperative agreements 273

274
How does PHS regulate conflict?
Regulations Require: Institution to act as “Enforcer” and “Reporter” Investigator to disclose “significant financial interests” 274

275
Institution as “Enforcer”
Must establish and enforce a written process that identifies, and manages , reduces, or eliminates any significant financial interests of an Investigator. Inform Investigator of process and reporting responsibilities. 42 CFR § 45 CFR § 94.4 275

276
Institution as “Reporter”
Must certify to PHS agency that written enforced COI process is in effect. Must report any COI (including Significant Financial Interest) to PHS agency and assure that the COI has been managed, reduced, or eliminated on an annual basis or as new reportable Significant Financial Interests are identified Must report to PHS agency any failure of an Investigator to comply with Institution’s COI process Must make COI information available to HHS 42 CFR § , 45 CFR § 94.4, 94.6 276
 to PHS agency and assure that the COI has been managed, reduced, or eliminated on an annual basis or as new reportable Significant Financial Interests are identified. Must report to PHS agency any failure of an Investigator to comply with Institution’s COI process. Must make COI information available to HHS. 42 CFR § , CFR § 94.4,")
277
Who must disclose a Significant Financial Interest?
Principal Investigator Any other person who is responsible for the design, conduct, or reporting of the PHS grant (e.g. Co-investigators). Their spouse and dependent children 42 CFR § 45 CFR § 94.3 277
. Their spouse and dependent children. 42 CFR § CFR §")
278
What is a Significant Financial Interest?
Anything of monetary value 42 CFR § 45 CFR § 94.3 278

279
Examples Significant Financial Interest?
Salary other payments of services (consulting fees, honoraria) Equity interests (stocks, stock options, any ownership interests) Intellectual Property Rights (patents, copyrights, and royalties) 42 CFR § 45 CFR § 94.3 279
 Equity interests. (stocks, stock options, any ownership interests) Intellectual Property Rights. (patents, copyrights, and royalties) 42 CFR § CFR §")
280
Examples Not Significant Financial Interest?
Salaries, royalties or other remuneration from the Institution Income from seminars, lectures, teaching engagements sponsored by public or non- profit entities Equity Interest that does not exceed $10,000 in value and does not represent more than 5% ownership interest in any single entity Salaries, royalties, or other payments that do not exceed $10,000 over a 12 month period 42 CFR § 45 CFR § 94.3 280

281
How does an Institution manage, reduce or eliminate COI?
Public Disclosure of SFI Monitoring of PHS research by independent reviewers Modification of the research plan Disqualification from participation in all or part of the PHS research Divestiture of significant financial interests Severance of relationships that create actual or potential conflicts 42 CFR § 45 CFR § 94.5 281

282
FDA Regulations (Clinical Trials)
282

283
Who is a Clinical Investigator?
Investigators or Sub-Investigators who are directly involved in the treatment or evaluation of research subjects Their spouse and dependent children 21 CFR § 54.2 283

284
What financial relationships and interests must be disclosed?
Compensation to the investigator for conducting the Study is based on the outcome of the Study Any significant payments of other sorts from Sponsor of monetary value of more than $25,000 (i.e. grant to fund other research, equipment, consulting fees, honoraria) Any proprietary interest in the Drug/Device Any significant equity interest in the Sponsor. - Publicly-held entity: Exceeds $50,000 - Privately-held entity: Cannot be readily determined Any steps taken to minimize the potential for bias resulting from any of the above arrangements, interests, or payments 21 CFR § 54.4 284
 Any proprietary interest in the Drug/Device. Any significant equity interest in the Sponsor. - Publicly-held entity: Exceeds $50, Privately-held entity: Cannot be readily determined. Any steps taken to minimize the potential for bias resulting from any of the above arrangements, interests, or payments. 21 CFR §")
285
FDA Review After the Study is completed, Sponsor will submit its IND/IDE application to the FDA which will contain Form 3454 (Certification) or Form 3455 (Disclosure) for each Clinical Investigator. If FDA determines that the disclosed financial interests raise a serious question about the integrity of the data, it may take any action necessary to ensure reliability of the data. 21 CFR § 54.5 42 CFR § 45 CFR § 94.6 285
 or Form 3455 (Disclosure) for each Clinical Investigator. If FDA determines that the disclosed financial interests raise a serious question about the integrity of the data, it may take any action necessary to ensure reliability of the data. 21 CFR § CFR § CFR §")
286
FDA Actions Audit of the data Requesting further analysis of data
Requesting further independent studies to confirm results Reject results 21 CFR § 54.5 286

287
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Protection of Human Subject (HIPAA) 287
 287.")
288
Federal Anti-Kickback Statute
Arrangement between vendor and healthcare professional Induces or influences the purchase, order, or referral of drugs, devices, products, services, or other items Reimbursable under a federal healthcare program 288

289
Federal Anti-Kickback Statute
Criminal Offense Penalties up to $25K or imprisonment up to 5 years, or both Exclusion from Federal healthcare programs (i.e. Medicare, Medicaid) HHS may seek civil penalty $50K for each act that violates the AKS 289
 HHS may seek civil penalty $50K for each act that violates the AKS")
290
Personal Services Safe Harbor (Application to Research Agreements)
Contract in writing and signed by parties Specify all services Term of contract over 1 year Compensation Set in advance FMV Not determined by volume, referrals, etc. 290

291
Relevance in Research? Research arrangement is suspect when:
Initiated/directed by Sponsor’s marketing dept Study results not shared with Sponsor’s science dept Duplicate research/ serves no legitimate purpose Product promotional activity masked as postmarket research Payments for services above FMV 291

292
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Protection of Human Subject (HIPAA) 292
 292.")
293
Federal False Claims Act
Organization knowingly submits a false claim for payment/approval to an agency of the federal government. “Knowingly” includes actual knowledge deliberate ignorance reckless disregard of the truth 293

294
Relates to Research? Certification Theory
Gov’t requires certifications: Apply for a grant Submit an application to the FDA Submit a claim for reimbursement for research-related care Whenever a false certification is made, FCA is violated. 294

295
Examples in Research False certification in grant applications
False statements in use of grant funds Failure to make financial disclosures (FDA Form 3454) Failure to make complete financial disclosures (FDA Form 3455) Allocations of costs in funded research Medicare/Medicaid billing Double-billing
 Failure to make complete financial disclosures (FDA Form 3455) Allocations of costs in funded research. Medicare/Medicaid billing. Double-billing.")
296
Federal False Claims Act
Civil action May include criminal penalties Damages plus $11K penalty for each false claim If brought by whistleblower, s/he may receive as much as 30% reward if government does not intervene Potential exclusion from federal healthcare programs 296

297
AKS and FCA Cases Warner-Lambert (FCA) $430 million
$83.6million for FCA Violation $106.4 million for State Settlement $240 million Federal Criminal Fine Whistleblower’s share = $24.64 million Corporate Integrity Agreement 297

298
AKS and FCA Cases TAP Pharmaceuticals (AKS & FCA) $875 million
$559.5 million for FCA Violations $25.5 million for State Settlement $290 million federal criminal fine (DOJ’s Crime Victims Fund ) Whistleblower share 1 = $78 million (TAP VP of Sales) Whistleblower share 2 = $17.1million (Tufts Health Plan & Physician) Corporate Integrity Agreement 298
 Whistleblower share 1 = $78 million (TAP VP of Sales) Whistleblower share 2 = $17.1million (Tufts Health Plan & Physician) Corporate Integrity Agreement")
299
AKS and FCA Cases Arizona Heart Institute (FCA) $6.7 Million
$5.8Million Arizona Heart Institute $900K Medical Practice Groups Corporate Integrity Agreement (5 years) Violated FCA by submitting Medicare claims for experimental procedures 299
 Violated FCA by submitting Medicare claims for experimental procedures")
300
ROAD MAP I General II Contracts Why have a Research Agreement?
Confidentiality Agreements Clinical Trial Agreements Government Grants/Cooperative Grants III Fraud & Abuse Regulations Conflict of Interest Regulations Federal Anti-Kickback Statute Federal False Claims Act IV Other Issues 300

301
Double-Billing Seeking multiple reimbursement for same services Seeking Medicare/Medicaid reimbursement for services reimbursed by sponsors/grant funds

302
HIPAA Informed Consent – Authorization Limited Data Set Agreements

303
Insider Trading Providing results to a third party for the purpose of assessing corporate stock violates insider trading laws Violates Confidentiality Agreements

Similar presentations




 is a review committee established to help protect the rights and welfare of human research subjects.>")




 An IRB shall conduct continuing review of research covered by this.>")

, Informed Consent, and Responsibilities Requirement for IRBs -DHHS: 45 CFR Part 46 -FDA: 21 CFR 56 Requirement for and.>")











