Download presentation
Presentation is loading. Please wait.

1
مهندسی ژنتیک

2
تعریف: دست ورزی مستقیم ژنوم یک موجود با استفاده از فن آوری دی ان ای نوترکیب این فرایند شامل انتقال ماده ژنتیکی به موجود هدف می باشد که در این حالت ارگانیسم دریافت کننده، GMO (genetically modified organism) نامیده می شود. Zebrafisch GloFish
 نامیده می شود. Zebrafisch. GloFish.")
3
مهندسی ژنتیک چیست؟ مهمترین بخش بیوتکنولوژی نوین
شناسایی، جداسازی، بریدن وچسباندن مولکول های DNA ایجاد DNA نوترکیب به طور کلی دستکاری ساختارهای ژنتیکی و خلق موجوداتی با ترکیب جدید ژنتیکی

4
تاریخچه 1970 شناسایی اولین آنزیم برشی توسط اسمیت Hind II))
1971 تهیه اولین نقشه برشی DNA حلقوی ویروس SV40 با استفاده از آنزیم فوق 1972 اولین تلاش ها برای ترکیب دو مولکول DNA از منشاء های مختلف توسط پائول برگ 1973 ادغام DNA ویروسی و باکتریایی در یک پلاسمید توسط استنلی کوهن و هربرت بویر و تولید اولین موجود حامل DNA نوترکیب 1977 تولید هورمون انسانی هوماتوستاتین توسط شرکت GenTech 1977 ابداع روش خاتمه زنجیره برای توالی یابی DNA توسط Frederick Sanger

5
تاریخچه 1978 تولید نخستین واکسن بر ضد هپاتیت B با استفاده از تکنیک DNA نوترکیب 1983 ابداع روش واکنش زنجیره ای پلیمراز (PCR) توسط Kary Mullis 1993 تولید هورمون رشد گاوی (BGH) در باکتری و خوراندن آن به گاوها برای افزایش تولید شیر آنها 1994 تولید گوجه فرنگی فلاور ساور ( (FlavrSavr با عمر انبارداری طولانی تر 1996 تولید بز تراریخت به نام گریس که دارای پروتئین ضد سرطان در شیر خود بود 1997 تولد اولین موجود کلون شده به نام دالی در اسکاتلند 1998 تا 2002 کلونینگ موجودات مختلف مثل مرغ، بز، میمون و گربه در اسکاتلند، کانادا و آمریکا
 در باکتری و خوراندن آن به گاوها برای افزایش تولید شیر آنها تولید گوجه فرنگی فلاور ساور ( (FlavrSavr با عمر انبارداری طولانی تر تولید بز تراریخت به نام گریس که دارای پروتئین ضد سرطان در شیر خود بود تولد اولین موجود کلون شده به نام دالی در اسکاتلند تا 2002 کلونینگ موجودات مختلف مثل مرغ، بز، میمون و گربه در اسکاتلند، کانادا و آمریکا.")
6
The most important topics:
Gene Isolation & DNA extraction Restriction Enzymes DNA (Gene) cloning PCR Plasmids & vectors Gene transfer Hosts
 cloning. PCR. Plasmids & vectors. Gene transfer. Hosts.")
7
DNA preparation The procedure for total DNA preparation from a culture of bacterial cells can be divided into four stages: A culture of bacteria is grown and then harvested. The cells are broken open to release their contents. This cell extract is treated to remove all components except the DNA. The resulting DNA solution is concentrated.

8
DNA Extraction 1- Breaking the cell walls, commonly referred to as cell disruption or cell lysis, to expose the DNA within. This is commonly achieved by chemical and physical methods-blending, grinding or sonicating the sample. 2- Removing membrane lipids by adding a detergent or surfactants. 3- Removing proteins by adding a protease (optional but almost always done). 4- Removing RNA by adding an RNase (often done). 5- Precipitating the DNA with an alcohol — usually ice-cold ethanol or isopropanol. Since DNA is insoluble in these alcohols, it will aggregate together, giving a pellet upon centrifugation. This step also removes alcohol-soluble salt.
. 4- Removing RNA by adding an RNase (often done). 5- Precipitating the DNA with an alcohol — usually ice-cold ethanol or isopropanol. Since DNA is insoluble in these alcohols, it will aggregate together, giving a pellet upon centrifugation. This step also removes alcohol-soluble salt.")
9
growth and harvest of bacteria

10
growth and harvest of bacteria

11
growth and harvest of bacteria

12
Preparation of a cell extract
The bacterial cell is enclosed in a cytoplasmic membrane and surrounded by a rigid cell wall. with E. coli and related organisms, weakening of the cell wall is usually brought about by lysozyme, ethylene diamine tetra acetate (EDTA),or a combination of both. usually a detergent such as sodium dodecyl sulphate (SDS) is also added.
,or a combination of both. usually a detergent such as sodium dodecyl sulphate (SDS) is also added.")
13
What does the detergent do?
Each cell is surrounded by a cell membrane DNA is in the nucleus of a cell, which is also surrounded by a membrane

14
Detergent: Breaks apart membranes by attaching to the lipids (fats) & proteins in the membranes
 & proteins in the membranes")
15
Preparation of a cell extract

16
Carbohydrates (sugars) DNA contaminants
The cell and nuclear membranes have been broken apart, as well as all of the organelle membranes,such as those around the mitochondria and chloroplasts. So what is left? Proteins RNA Carbohydrates (sugars) DNA contaminants
 DNA. contaminants.")
17
Purification of DNA from a cell extract
Degradation of contaminants Separation of DNA from contaminants

18
Purification of DNA from a cell extract
The standard way to deproteinize a cell extract is to add phenol or a 1 : 1 mixture of phenol and chloroform. These organic solvents precipitate proteins but leave the nucleic acids (DNA and RNA) in aqueous solution. In some case treat the cell extract with a protease such as pronase or proteinase K before phenol extraction. Some RNA molecules, especially messenger RNA (mRNA), are removed by phenol treatment, but most remain with the DNA in the aqueous layer. The only effective way to remove the RNA is with the enzyme ribonuclease, which rapidly degrades these molecules into ribonucleotide subunits.
 in aqueous solution. In some case treat the cell extract with a protease such as pronase or proteinase K before phenol extraction. Some RNA molecules, especially messenger RNA (mRNA), are removed by phenol treatment, but most remain with the DNA in the aqueous layer. The only effective way to remove the RNA is with the enzyme ribonuclease, which rapidly degrades these molecules into ribonucleotide subunits.")
20
Using ion-exchange chromatography to purify DNA from a cell extract

21
Concentration of DNA samples
Collecting DNA by ethanol precipitation. (b) For less concentrated solutions ethanol is added (at a ratio of 2.5 volumes of absolute ethanol to 1 volume of DNA solution) and precipitated DNA collected by centrifugation. (a) Absolute ethanol is layered on top of a concentrated solution of DNA. Fibers of DNA can be withdrawn with a glass rod.
 For less concentrated solutions ethanol is added (at a ratio of 2.5 volumes of absolute ethanol to 1 volume of DNA solution) and precipitated DNA collected by centrifugation. (a) Absolute ethanol is layered on top of a concentrated solution of DNA. Fibers of DNA can be withdrawn with a glass rod.")
22
Other methods for the preparation of total cell DNA )The CTAB method(
Plant tissues are particularly difficult in this respect as they often contain large amounts of carbohydrates that are not removed by phenol extraction. One method makes use of a detergent called cetyl trimethyl ammonium bromide (CTAB), which forms an insoluble complex with nucleic acids. When CTAB is added to a plant cell extract the nucleic acid–CTAB complex precipitates, leaving carbohydrate, protein, and other contaminants in the supernatant. The precipitate is then collected by centrifugation and resuspended in 1 M sodium chloride, which causes the complex to break down. The nucleic acids can now be concentrated by ethanol precipitation and the RNA removed by ribonuclease treatment.
, which forms an insoluble complex with nucleic acids. When CTAB is added to a plant cell extract the nucleic acid–CTAB complex precipitates, leaving carbohydrate, protein, and other contaminants in the supernatant. The precipitate is then collected by centrifugation and resuspended in 1 M sodium chloride, which causes the complex to break down. The nucleic acids can now be concentrated by ethanol precipitation and the RNA removed by ribonuclease treatment.")
23
Preparation of plasmid DNA
separation of plasmid DNA from the large amount of bacterial chromosomal DNA Separation on the basis of size Separation on the basis of conformation

24
Separation on the basis of size

25
Separation on the basis of conformation

26
Ethidium bromide–caesium chloride density gradient centrifugation
Purification of plasmid DNA by EtBr–CsCl density gradient centrifugation

27
Gel Electrophoresis DNA is placed at one end of a gel
A current is applied to the gel DNA molecules are negatively charged and move toward positive end of gel Smaller molecules move faster than larger ones

29
DNA loading bp

32
Gel Electrophoresis

33
gel documentation systems

36
The influence of DNA size on migration rate during conventional gel electrophoresis

37
The orthogonal field alternation gel electrophoresis (OFAGE)
(a) Conventional agarose gel electrophoresis (b) OFAGE
 Conventional agarose gel electrophoresis. (b) OFAGE.")
38
Manipulation of Purified DNA

39
Manipulation of Purified DNA
Almost all DNA manipulative techniques make use of purified enzymes Within the cell these enzymes participate in essential processes such as DNA replication and transcription, breakdown of unwanted or foreign DNA (e.g., invading virus DNA), repair of mutated DNA, and recombination between different DNA molecules
, repair of mutated DNA, and recombination between different DNA molecules.")
40
The range of DNA manipulative enzymes
DNA manipulative enzymes can be grouped into four broad classes, depending on the type of reaction that they catalyze: Nucleases are enzymes that cut, shorten, or degrade nucleic acid molecules Ligases join nucleic acid molecules together Polymerases make copies of molecules Modifying enzymes remove or add chemical groups

41
Nucleases Nucleases degrade DNA molecules by breaking the phosphodiester bonds that link one nucleotide to the next in a DNA strand. There are two different kinds of nuclease: Exonucleases remove nucleotides one at a time from the end of a DNA molecule. Endonucleases are able to break internal phosphodiester bonds within a DNA molecule.

42
An exonuclease

43
Two type of exonuclease

44
An endonuclease

45
Two type of endonuclease
(a) S1 nuclease, which cleaves only single- stranded DNA, including single-stranded nicks in mainly double-stranded molecules. (b) DNase I,which cleaves both single- and double-stranded DNA.
 S1 nuclease, which cleaves only single- stranded DNA, including single-stranded nicks in mainly double-stranded molecules. (b) DNase I,which cleaves both single- and. double-stranded DNA.")
46
Ligases (a) Repair of a discontinuity—a missing phosphodiester bond in one strand of a double-stranded molecule. (b) Joining two molecules together.
 Joining two molecules together.")
47
Polymerases DNA polymerases are enzymes that synthesize a new strand of DNA complementary to an existing DNA or RNA template (a) The basic reaction: a new DNA strand is synthesized in the 5′ to 3′ direction.
 The basic reaction: a new DNA strand is synthesized in the 5′ to 3′ direction.")
48
DNA polymerase I (b) DNA polymerase I, which initially fills in nicks but then continues to synthesize a new strand, degrading the existing one as it proceeds.
 DNA polymerase I, which initially fills in nicks but then continues to synthesize a new strand, degrading the existing one as it proceeds.")
49
The Klenow fragment (c) The Klenow fragment, which only fills in nicks.
 The Klenow fragment, which only fills in nicks.")
50
Reverse transcriptase
The ability of this enzyme to synthesize a DNA strand complementary to an RNA template is central to the technique called complementary DNA (cDNA) cloning. (d) Reverse transcriptase, which uses a template of RNA.
 cloning. (d) Reverse transcriptase, which uses a template of RNA.")
51
The Taq DNA polymerase The Taq DNA polymerase used in the polymerase chain reaction (PCR) is the DNA polymerase I enzyme of the bacterium Thermus aquaticus.
 is the DNA polymerase I enzyme of the bacterium Thermus aquaticus.")
52
DNA modifying enzymes There are numerous enzymes that modify DNA molecules by addition or removal of specific chemical groups. The most important are as follows: Alkaline phosphatase Polynucleotide kinase Terminal deoxynucleotidyl transferase

53
Alkaline phosphatase Alkaline phosphatase (from E. coli, calf intestinal tissue, or arctic shrimp), which removes the phosphate group present at the 5′ terminus of a DNA molecule.
, which removes the phosphate group present at the 5′ terminus of a DNA molecule.")
54
Polynucleotide kinase
Polynucleotide kinase (from E. coli infected with T4 phage), which has the reverse effect to alkaline phosphatase, adding phosphate groups onto free 5′ termini.
, which has the reverse effect to alkaline phosphatase, adding phosphate groups onto free 5′ termini.")
55
Terminal deoxynucleotidyl transferase
Terminal deoxynucleotidyl transferase (from calf thymus tissue), which adds one or more deoxyribonucleotides onto the 3′ terminus of a DNA molecule.
, which adds one or more deoxyribonucleotides onto the 3′ terminus of a DNA molecule.")
56
Restriction endonucleases
Gene cloning requires that DNA molecules be cut in a very precise and reproducible fashion.

57
The discovery and function of restriction endonucleases
The initial observation that led to the eventual discovery of restriction endonucleases was made in the early 1950s, when it was shown that some strains of bacteria are immune to bacteriophage infection, a phenomenon referred to as host- controlled restriction. over 2500 different ones have been isolated and more than 300 are available for use in the laboratory.

58
The function of a restriction endonuclease in a bacterial cell
(a) phage DNA is cleaved, but (b) bacterial DNA is not.
 phage DNA is cleaved, but (b) bacterial DNA is not.")
59
Classification of restriction endonuclease
Three different classes of restriction endonuclease are recognized: Types I and III are rather complex and have only a limited role in genetic engineering. Type II restriction endonucleases, on the other hand, are the cutting enzymes that are so important in gene cloning.

60
The recognition sequences for some of the most frequently used restriction endonucleases

61
Blunt ends and sticky ends

62
Isoschizomers ; Neoschizomers; Isocaudomers
Restriction enzymes that are isolated from different organisms and recognize identical sequences are called isoschizomers. Neoschizomers Isoschizomeric enzymes that possess different cleavage sites in a particular recognition sequence are called neoschizomers (e.g., Sma I [CCC↓GGG] and XmaC I [C↓CCGGG]). Isocaudomers are pairs of restriction enzymes that have slightly different recognition sequences but upon cleavage generate identical termini. For example the enzymes Mbo I and BamH I are isocaudomers:
. Isocaudomers. are pairs of restriction enzymes that have slightly different recognition sequences but upon cleavage generate identical termini. For example the enzymes Mbo I and BamH I are isocaudomers:")
63
Isocaudomers The same sticky ends produced by different restriction endonucleases

64
The frequency of recognition sequences in a DNA molecule
The number of recognition sequences for a particular restriction endonuclease in a DNA molecule of known length can be calculated mathematically. A tetranucleotide sequence(e.g., GATC) should occur once every 44 = 256 nucleotides, and a hexanucleotide (e.g., GGATCC) once every 46 = nucleotides. These calculations assume that the nucleotides are ordered in a random fashion four different nucleotides are present in equal proportions (i.e., the GC content = 50%).
 should occur once every 44 = 256 nucleotides, and a hexanucleotide (e.g., GGATCC) once every 46 = 4096 nucleotides. These calculations assume that. the nucleotides are ordered in a random fashion. four different nucleotides are present in equal proportions (i.e., the GC content = 50%).")
65
Ligation – joining DNA molecules together
The final step in construction of a recombinant DNA molecule is the joining together of the vector molecule and the DNA to be cloned

66
Sticky ends increase the efficiency of ligation
Ligation of complementary sticky ends is much more efficient. This is because compatible sticky ends can base pair with one another by hydrogen bonding forming a relatively stable structure for the enzyme to work on. Putting sticky ends onto a blunt-ended molecule: Linkers Adaptors Producing sticky ends by homopolymer tailing

67
Linkers

68
The Linkers potential drawback

69
Adaptors Adaptors, like linkers, are short synthetic oligonucleotides. But unlike linkers, an adaptor is synthesized so that it already has one sticky end.

70
Adaptors

72
Producing sticky ends by homopolymer tailing
Tailing involves using the enzyme terminal deoxynucleotidyl transferase to add a series of nucleotides onto the 3′-OH termini of a double-stranded DNA molecule. If this reaction is carried out in the presence of just one deoxyribonucleotide, a homopolymer tail is produced.

73
homopolymer tailing Frequently polydeoxycytosine (poly(dC)) tails are attached to the vector and poly(dG) to the DNA to be cloned.
) tails are attached to the vector and poly(dG) to the DNA to be cloned.")
74
Repair of the recombinant DNA molecule
In practice, the poly(dG) and poly(dC) tails are not usually exactly the same length, and the base-paired recombinant molecules that result have nicks as well as discontinuities. Repair is therefore a two-step process: Using Klenow polymerase to fill in the nicks Using DNA ligase to synthesize the final phosphodiester bonds.
 and poly(dC) tails are not usually exactly the same length, and the base-paired recombinant molecules that result have nicks as well as discontinuities. Repair is therefore a two-step process: Using Klenow polymerase to fill in the nicks. Using DNA ligase to synthesize the final phosphodiester bonds.")
76
Transformation

77
Gene transfer: Horizontal Vertical

78
Horizontal Gene transfer in bacteria
There are three processes by which exogenous genetic material may be introduced into a bacterial cell: Transformation (Capturing DNA from solution) Transduction (Phage-mediated) Conjugation (Bacterial Sex)
 Transduction. (Phage-mediated) Conjugation. (Bacterial Sex)")
79
transformation In molecular biology transformation is the genetic alteration of a cell resulting from the direct uptake, incorporation and expression of exogenous genetic material (exogenous DNA) from its surroundings and taken up through the cell membrane(s).
 from its surroundings and taken up through the cell membrane(s).")
80
Transformation was first demonstrated in 1928 by British bacteriologist Frederick Griffith.
Frederick Griffith (1879 - 1941) British physician, pathologist, bacteriologist Known for discovery of pneumococcal transformation
 British physician, pathologist, bacteriologist Known for discovery of pneumococcal transformation.")
81
Griffith's experiment

82
DNA or Protein? Which one is genetic material?

83
Avery, MacLeod and McCarty, in 1944 discovered that DNA is the material of which genes and chromosomes are made. Oswald Theodore Avery ForMemRS (October 21, 1877 – February 2, 1955) was a Canadian-born American physician and medical researcher. Maclyn McCarty (South Bend, Indiana, June 9, – January 2, 2005) was an American geneticist. Colin Munro MacLeod (January 28, 1909 — February 11, 1972) was a Canadian-American geneticist.
 was a Canadian-born American physician and medical researcher. Maclyn McCarty (South Bend, Indiana, June 9, 1911 – January 2, 2005) was an American geneticist. Colin Munro MacLeod (January 28, 1909 — February 11, 1972) was a Canadian-American geneticist.")
84
Avery–MacLeod–McCarty experiment
Chemical analysis showed that the proportions of carbon, hydrogen, nitrogen, and phosphorus in this active portion were consistent with the chemical composition of DNA. To show that it was DNA rather than some small amount of RNA, protein, or some other cell component that was responsible for transformation, Avery and his colleagues used a number of biochemical tests. They found that trypsin, chymotrypsin and ribonuclease (enzymes that break apart proteins or RNA) did not affect it, but an enzyme preparation of "deoxyribonucleodepolymerase" (a crude preparation, obtainable from a number of animal sources, that could break down DNA) destroyed the extract's transforming power
 did not affect it, but an enzyme preparation of deoxyribonucleodepolymerase (a crude preparation, obtainable from a number of animal sources, that could break down DNA) destroyed the extract s transforming power.")
85
The Hershey–Chase experiments in 1952 helped to confirm that DNA was the genetic material.
Martha Cowles Chase (November 30, 1927 – August 8, 2003 was an American geneticist and Alfred Hershey’s laboratory assistant Alfred Day Hershey (December 4, 1908 – May 22, 1997) was an American Nobel Prize-winning bacteriologist and geneticist.
 was an American Nobel Prize-winning bacteriologist and geneticist.")
86
Hershey–Chase experiment
Hershey and Chase were testing two competing hypotheses: 1- DNA is the genetic material 2- protein is the genetic material DNA was radiolabeled with radioactive phosphorus-32 (32P) Protein was radiolabeled with radioactive sulfur-35 (35S)
 Protein was radiolabeled with radioactive sulfur-35 (35S)")
87
Hershey–Chase experiment
radioactive sulfur-35 (35S) tracer found in the supernatant with the phage coats DNA is genetic material
 tracer found in the supernatant with the phage coats. DNA. is genetic material.")
88
Transformation: uptake of naked DNA (DNA without associated cells or proteins) by competent bacteria competent : competence is the ability of a cell to take up extracellular ("naked") DNA from its environment. natural competence induced or artificial competence
 DNA from its environment. natural competence. induced or artificial competence.")
89
Artificial competence
Use of divalent cations most commonly calcium chloride (CaCl2) solution under cold condition, and then exposed to a pulse of heat shock. Electroporation In this method the cells are briefly shocked with an electric field of kV/cm which is thought to create holes in the cell membrane through which the plasmid DNA may enter. After the electric shock the holes are rapidly closed by the cell's membrane-repair mechanisms.
 solution under cold condition, and then exposed to a pulse of heat shock. Electroporation. In this method the cells are briefly shocked with an electric field of kV/cm which is thought to create holes in the cell membrane through which the plasmid DNA may enter. After the electric shock the holes are rapidly closed by the cell s membrane-repair mechanisms.")
90
Polymerase Chain Reaction (PCR)
A scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA Developed in 1983 by Kary Mullis Kary Mullis wined Nobel Prize in Chemistry in 1993 Kary Banks Mullis (born December 28,1944) Nobel Prize winning American biochemist.
 Nobel Prize winning American biochemist.")
91
PCR - Applications DNA cloning Forensic medicine In-vitro mutation
DNA sequencing Gene identification Molecular phylogenetics Detection of disease mutations Gene expression studies 22

96
pcr Schematic drawing of the PCR cycle. Denaturing at 96°C. 5 min
Annealing at 62°C min Extension at 72°C min Final extension at 72°C min Storage at 4°C min 30 x

97
pcr

98
Number of short products synthesized after 25 cycles of PCR with different numbers of starting molecule. Note: The numbers assume that amplification is 100% efficient, none of the reactants becoming limiting during the course of the PCR.

99
PCR components A basic PCR set up requires several components and reagents. These components include: DNA template Two primers: reverse & forward Taq DNA polymerase* dNTPs (Deoxynucleoside triphosphates : dATP, dGTP, dCTP, dTTP) Buffer solution MgCl2 (Divalent cations, magnesium or manganese ions; generally Mg2+ is used,) dd water * Taq polymerase is a thermostable DNA polymerase named after the thermophilic bacterium Thermus aquaticus from which it was originally isolated by Thomas D. Brock in 1965.
 Buffer solution. MgCl2 (Divalent cations, magnesium or manganese ions; generally Mg2+ is used,) dd water. * Taq polymerase is a thermostable DNA polymerase named after the thermophilic bacterium Thermus aquaticus from which it was originally isolated by Thomas D. Brock in")
100
A thermal cycler for PCR

101
An older model three-temperature thermal cycler for PCR

102
A thermal cycler for PCR

103
annealing temperature
The ideal annealing temperature must be : low enough to enable hybridization between primer and template high enough to prevent mismatched hybrids from forming This temperature can be estimated by determining the melting temperature or Tm of the primer–template hybrid. The Tm is the temperature at which the correctly base-paired hybrid dissociates (“melts”). A temperature 1–2°C below this should be low enough to allow the correct primer–template hybrid to form, but too high for a hybrid with a single mismatch to be stable. The Tm can be determined experimentally but is more usually calculated from the simple formula: Tm = (4 × [G + C]) + (2 × [A + T])°C
. A temperature 1–2°C below this should be low enough to allow the correct primer–template hybrid to form, but too high for a hybrid with a single mismatch to be stable. The Tm can be determined experimentally but is more usually calculated from the simple formula: Tm = (4 × [G + C]) + (2 × [A + T])°C.")
104
Calculating the Tm of a primer

105
Variants of PCR Probe Amplification (MLPA) Multiplex-PCR
Nested PCR Splicing by overlap extension (SOEing PCR) Real time PCR (Quantitative PCR (Q-PCR)) Reverse Transcription PCR (RT-PCR) Solid Phase PCR Thermal asymmetric interlaced PCR (TAIL-PCR) Touchdown PCR (Step-down PCR) PAN-AC Universal Fast Walking Allele-specific PCR Assembly PCR Asymmetric PCR Helicase-dependent amplification Hot start PCR Intersequence-specific PCR Inverse PCR Ligation-mediated PCR Methylation-specific PCR (MSP) Miniprimer PCR Multiplex Ligation-dependent
 Real time PCR (Quantitative PCR (Q-PCR)) Reverse Transcription PCR (RT-PCR) Solid Phase PCR. Thermal asymmetric interlaced PCR (TAIL-PCR) Touchdown PCR (Step-down PCR) PAN-AC. Universal Fast Walking. Allele-specific PCR. Assembly PCR Asymmetric PCR. Helicase-dependent amplification. Hot start PCR. Intersequence-specific PCR. Inverse PCR. Ligation-mediated PCR. Methylation-specific PCR (MSP) Miniprimer PCR. Multiplex Ligation-dependent.")
106
PCR - Advantages Speed Ease of use Sensitive 20

107
PCR - Disadvantages Need sequence information
Limited amplification size Limited amounts of product Infidelity of DNA replication 21

108
The error rate of Taq polymerase
The error rate has been estimated at one mistake for every 9000 nucleotides of DNA that is synthesized, which might appear to be almost insignificant but which translates to one error in every 300 bp for the PCR products obtained after 30 cycles.

109
Inverse PCR

110
Nested PCR Nested PCR is a variation of the polymerase chain reaction (PCR), in that two pairs (instead of one pair) of PCR primers are used to amplify a fragment. The first pair of PCR primers amplify a fragment similar to a standard PCR. However, a second pair of primers called nested primers (as they lie / are nested within the first fragment) bind inside the first PCR product fragment to allow amplification of a second PCR product which is shorter than the first one. The advantage of nested PCR is that if the wrong PCR fragment was amplified, the probability is quite low that the region would be amplified a second time by the second set of primers. Thus, Nested PCR is a very specific PCR amplification.
, in that two pairs (instead of one pair) of PCR primers are used to amplify a fragment. The first pair of PCR primers amplify a fragment similar to a standard PCR. However, a second pair of primers called nested primers (as they lie / are nested within the first fragment) bind inside the first PCR product fragment to allow amplification of a second PCR product which is shorter than the first one. The advantage of nested PCR is that if the wrong PCR fragment was amplified, the probability is quite low that the region would be amplified a second time by the second set of primers. Thus, Nested PCR is a very specific PCR amplification.")
112
Nested PCR Target DNA two pairs of PCR primers are use:

113
SOEing PCR

114
SOEing PCR

115
Real time pcr TaqMan® Probes Molecular Beacons Scorpions® FRET Probes
SYBR® Green Assay High Resolution Melting Analysis (HRMA) Dual Labeled Probe: TaqMan® Probes Molecular Beacons Scorpions® FRET Probes
![]()
116
High Resolution Melting (HRM)
Mechanism High Resolution Melting Analysis (HRM) is a post PCR method. The region of interest within the DNA sequence is first amplified using the polymerase chain reaction. During this process, special saturation dyes are added to the reaction, that fluoresce only in the presence of double stranded DNA. Such dyes are known as Intercalating dyes. During PCR, the region of interest amplified is known as Amplicon. As the amplicon concentration in the reaction tube increases the fluorescence exhibited by the double stranded amplified product also increases. After the PCR process the HRM analysis begins. In this process the amplicon DNA is heated gradually from around 50°C up to around 95°C. As the temperature increases, at a point the melting temperature of the amplicon is reached and the sample DNA denatures and the double stranded DNA melts apart. Due to this the fluorescence fades away. This is because in the absence of double stranded DNA the intercalating dyes have nothing to bind to and they only fluoresce at a low level. This observation is plotted showing the level of fluorescence vs the temperature, generating a Melting Curve. Even a single base change in the sample DNA sequence causes differences in the HRM curve. Since different genetic sequences melt at slightly different rates, they can be viewed, compared, and detected using these curves.
 is a post PCR method. The region of interest within the DNA sequence is first amplified using the polymerase chain reaction. During this process, special saturation dyes are added to the reaction, that fluoresce only in the presence of double stranded DNA. Such dyes are known as Intercalating dyes. During PCR, the region of interest amplified is known as Amplicon. As the amplicon concentration in the reaction tube increases the fluorescence exhibited by the double stranded amplified product also increases. After the PCR process the HRM analysis begins. In this process the amplicon DNA is heated gradually from around 50°C up to around 95°C. As the temperature increases, at a point the melting temperature of the amplicon is reached and the sample DNA denatures and the double stranded DNA melts apart. Due to this the fluorescence fades away. This is because in the absence of double stranded DNA the intercalating dyes have nothing to bind to and they only fluoresce at a low level. This observation is plotted showing the level of fluorescence vs the temperature, generating a Melting Curve. Even a single base change in the sample DNA sequence causes differences in the HRM curve. Since different genetic sequences melt at slightly different rates, they can be viewed, compared, and detected using these curves.")
117
High Resolution Melting (HRM)
")
118
Advantages of HRM Cost Effective High Resolution DNA Melt Analysis is cost effective than other genotyping technologies such as sequencing and Taqman SNP typing. This makes it ideal for large scale genotyping projects. Simple and Fast Work flow After performing PCR amplification of the target sequence, no additional instrumentation is required for HRM Analysis. Fast and Powerful HRM Analysis is fast and powerful, due to which it is able to accurately genotype huge number of samples in a short time, with a high level of accuracy. Low Reagent Consumption High Resolution Melting Analysis requires only PCR reaction volume for analysis of each sample, eliminating the need for HPLC solvents or Denaturing Gradient Gel Electrophoresis (DGGE) gels.
 gels.")
119
Applications of HRM SNP Genotyping DNA Mapping Mutation Scanning
Species Identification Zygosity testing DNA Methylation Analysis DNA Fingerprinting

120
TaqMan® probe functioning
While carrying out a TaqMan® experiment, a fluorogenic probe, complementary to the target sequence is added to the PCR reaction mixture. This probe is an oligonucleotide with a reporter dye attached to the 5' end and a quencher dye attached to the 3' end. Till the time the probe is not hydrolized, the quencher and the fluorophore remain in proximity to each other, separated only by the length of the probe. This proximity however, does not completely quench the flourescence of the reporter dye and a background flourescence is observed. During PCR, the probe anneals specifically between the forward and reverse primer to an internal region of the PCR product. The polymerase then carries out the extension of the primer and replicates the template to which the TaqMan® is bound. The 5' exonuclease activity of the polymerase cleaves the probe, releasing the reporter molecule away from the close vicinity of the quencher. The fluorescence intensity of the reporter dye, as a result increases. This process repeats in every cycle and does not interfere with the accumulation of PCR product.

121
TaqMan® Probes

122
TaqMan® probe applications:
1. Quantitative real time PCR. 2. DNA copy number measurements. 3. Bacterial identification assays. 4. SNP genotyping. 5. Verification of microarray results.

123
Molecular Beacons Introduction to Molecular Beacons Molecular beacons are single stranded hairpin shaped oligonucleotide probes. In the presence of the target sequence, they unfold, bind and fluoresce. The molecular beacon chemistry is one of the chemistries used to carry out a real time experiment.
![]()
124
Molecular Beacons Structure
A molecular beacon consists of 4 parts, namely: Loop: This is the base pair region of the molecular beacon which is complementary to the target sequence. Stem: The beacon stem sequence lies on both the ends of the loop. It is typically 5-7 bp long at the sequences at both the ends are complementary to each other. 5' fluorophore: Towards the 3' end of the molecular beacon, is attached a dye that fluoresces in presence of a complementary target. 3' quencher (non fluorescent): The quencher dye is covalently attached to the 3' end of the molecular beacon and when the beacon is in closed loop shape, prevents the fluorophore from emitting light.
![]()
125
Applications of Molecular Beacons
SNP detection Real-time nucleic acid detection Real-time PCR quantification Allelic discrimination and identification Multiplex PCR assays Diagnostic clinical assays
![]()
126
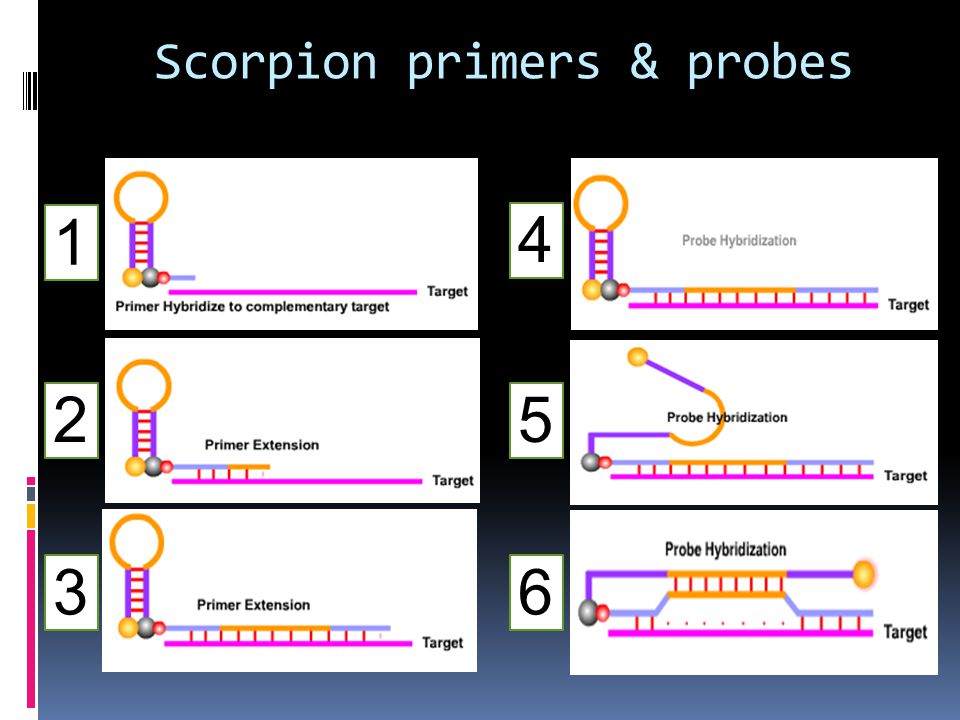
Scorpion primers & probes
Structure of the Scorpion primer The Scorpion primer carries a Scorpion probe element at the 5' end. The probe is a self- complementary stem sequence with a fluorophore at one end and a quencher at the other. The Scorpion primer sequence is modified at the 5'end. It contains a PCR blocker at the start of the hairpin loop (Usually HEG monomers are added as blocking agent). In the initial PCR cycles, the primer hybridizes to the target and extension occurs due to the action of polymerase. Scorpion primers can be used to examine and identify point mutations by using multiple probes. Each probe can be tagged with a different fluorophore to produce different colors. Scorpion® Primer functioning In Scorpion primers, the probe is physically coupled to the primer which means that the reaction leading to signal generation is a unimolecular one. This is in contrast to the bi- molecular collisions required by other technologies such as TaqMan® or Molecular Beacons. After one cycle of PCR extension completes, the newly synthesized target region will be attached to the same strand as the probe. Following the second cycle of denaturation and annealing, the probe and the target hybridize. The denaturation of the hairpin loop requires less energy than the new DNA duplex produced. Consequently, the hairpin sequence hybridizes to a part of the newly produced PCR product. This results in the separation of the fluorophore from the quencher and causes emission.
. In the initial PCR cycles, the primer hybridizes to the target and extension occurs due to the action of polymerase. Scorpion primers can be used to examine and identify point mutations by using multiple probes. Each probe can be tagged with a different fluorophore to produce different colors. Scorpion® Primer functioning. In Scorpion primers, the probe is physically coupled to the primer which means that the reaction leading to signal generation is a unimolecular one. This is in contrast to the bi- molecular collisions required by other technologies such as TaqMan® or Molecular Beacons. After one cycle of PCR extension completes, the newly synthesized target region will be attached to the same strand as the probe. Following the second cycle of denaturation and annealing, the probe and the target hybridize. The denaturation of the hairpin loop requires less energy than the new DNA duplex produced. Consequently, the hairpin sequence hybridizes to a part of the newly produced PCR product. This results in the separation of the fluorophore from the quencher and causes emission.")
127
Scorpion primers & probes structure

128
Scorpion primers & probes
1 4 2 5 3 6
129
Scorpion primers & probes

130
Fluorescence resonance energy transfer (FRET)
FRET probes are a pair of fluorescent probes placed in close proximity. Fluorophores are so chosen that the emission spectrum of one overlaps significantly with the excitation spectrum of the other. During FRET, the donor fluorophore excited by a light source, transfers its energy to an acceptor fluorophore when positioned in the direct vicinity of the former. The acceptor fluorophore emits light of a longer wavelength, which is detected in specific channels. The light source cannot excite the acceptor dye. FRET probes functioning The hybridization probe system consists of two oligonucleotides labeled with fluorescent dyes. The hybridization probe pair is designed to hybridize to adjacent regions on the target DNA. Each probe is labeled with a different marker dye. Interaction of the two dyes can only occur when both are bound to their target. The donor probe is labeled with fluorophore at the 3' end and the acceptor probe at 5' end. During PCR, the two different oligonucleotides hybridize to adjacent regions of the target DNA such that the fluorophores, which are coupled to the oligonucleotides, are in close proximity in the hybrid structure. The donor fluorophore (F1) is excited by an external light source, then passes part of its excitation energy to the adjacent acceptor fluorophore (F2). The excited acceptor fluorophore (F2) emits light at a different wavelength which can then be detected and measured.
 is excited by an external light source, then passes part of its excitation energy to the adjacent acceptor fluorophore (F2). The excited acceptor fluorophore (F2) emits light at a different wavelength which can then be detected and measured.")
133
Applications of FRET probes Quantitative PCR
DNA copy number measurements. Pathogen detection assays. SNP genotyping. Verification of microarray results.

134
Bisulfite treatment of DNA

136
Methylation-specific PCR (MSP)
")
137
vectors a vector is a DNA molecule used as a vehicle to transfer foreign genetic material into another cell. The four major types of vectors are: plasmids can hold up to 10 kb viruses (Viral vectors) Cosmids can hold up to 30 kb artificial chromosomes (BAC, YAC) bacs are able to hold up to 350 kb of DNA
 Cosmids can hold up to 30 kb. artificial chromosomes (BAC, YAC) bacs are able to hold up to 350 kb of DNA")
138
vectors Cloning vectors Expression vectors

139
plasmids

140
plasmids a plasmid is a double-stranded DNA molecule, in many cases, circular Plasmids usually occur naturally in bacteria, but are sometimes found in eukaryotic organisms (e.g., the 2-micrometre ring in Saccharomyces cerevisiae). Plasmid sizes: vary from 1 to over 1,000 kbp. Copy number: in a single cell can range anywhere from one to thousands under some circumstances. Rather, plasmids provide a mechanism for horizontal gene transfer within a population of microbes and typically provide a selective advantage under a given environmental state. a plasmid can contain inserts of only about 1–10 kbp
. Plasmid sizes: vary from 1 to over 1,000 kbp. Copy number: in a single cell can range anywhere from one to thousands under some circumstances. Rather, plasmids provide a mechanism for horizontal gene transfer within a population of microbes and typically provide a selective advantage under a given environmental state. a plasmid can contain inserts of only about 1–10 kbp.")
141
Plasmid types Another way to classify plasmids is by function. There are five main classes: Fertility F-plasmids which contain tra genes. They are capable of conjugation. Resistance (R)plasmids which contain genes that can build a resistance against antibiotics or poisons and help bacteria produce pili. Historically known as R-factors, before the nature of plasmids was understood. Col plasmids which contain genes that code for bacteriocins, proteins that can kill other bacteria. Degradative plasmids which enable the digestion of unusual substances, e.g. toluene and salicylic acid. Virulence plasmids which turn the bacterium into a pathogen. Plasmids can belong to more than one of these functional groups.
plasmids. which contain genes that can build a resistance against antibiotics or poisons and help bacteria produce pili. Historically known as R-factors, before the nature of plasmids was understood. Col plasmids. which contain genes that code for bacteriocins, proteins that can kill other bacteria. Degradative plasmids. which enable the digestion of unusual substances, e.g. toluene and salicylic acid. Virulence plasmids. which turn the bacterium into a pathogen. Plasmids can belong to more than one of these functional groups.")
142
Plasmid DNA may appear in one of five conformations in a gel:
Nicked open-circular DNA has one strand cut. Relaxed circular DNA is fully intact with both strands uncut, but has been enzymatically relaxed (supercoils removed). This can be modeled by letting a twisted extension cord unwind and relax and then plugging it into itself. Linear DNA has free ends, either because both strands have been cut or because the DNA was linear in vivo. This can be modeled with an electrical extension cord that is not plugged into itself. Supercoiled (or covalently closed-circular) DNA is fully intact with both strands uncut, and with an integral twist, resulting in a compact form. This can be modeled by twisting an extension cord and then plugging it into itself. Supercoiled denatured DNA is like supercoiled DNA, but has unpaired regions that make it slightly less compact; this can result from excessive alkalinity during plasmid preparation. slow fast
. This can be modeled by letting a twisted extension cord unwind and relax and then plugging it into itself. Linear DNA has free ends, either because both strands have been cut or because the DNA was linear in vivo. This can be modeled with an electrical extension cord that is not plugged into itself. Supercoiled (or covalently closed-circular) DNA is fully intact with both strands uncut, and with an integral twist, resulting in a compact form. This can be modeled by twisting an extension cord and then plugging it into itself. Supercoiled denatured DNA is like supercoiled DNA, but has unpaired regions that make it slightly less compact; this can result from excessive alkalinity during plasmid preparation. slow. fast.")
144
Plasmids containing: Origin of replication
Multiple cloning site (MCS, or polylinker) which is a short region containing several commonly used restriction sites allowing the easy insertion of DNA fragments at this location Selectable marker gene A selectable marker is a gene introduced into a cell, especially a bacterium or to cells in culture, that confers a trait suitable for artificial selection. Selectable markers are often antibiotic resistance genes
 which is a short region containing several commonly used restriction sites allowing the easy insertion of DNA fragments at this location. Selectable marker gene. A selectable marker is a gene introduced into a cell, especially a bacterium or to cells in culture, that confers a trait suitable for artificial selection. Selectable markers are often antibiotic resistance genes.")
145
Replication strategies for (a) a non-integrative plasmid, and (b) an episome.
 a non-integrative plasmid, and (b) an episome.")
146
Sizes of representative plasmids.

147
pUC18/19 vector

148
Multiple cloning site (MCS, or polylinker)
")
149
Incompatibility groups
Several different kinds of plasmid may be found in a single cell. In fact, cells of E. coli have been known to contain up to seven different plasmids at once. incompatibility is due to the sharing of one or more elements of the plasmid replication or partitioning systems Incompatibility may be: symmetric (either coresident plasmid is lost with equal probability) vectorial (one plasmid is lost exclusively or with higher probability than the other)
 vectorial. (one plasmid is lost exclusively or with higher probability than the other)")
150
Some cloning vectors TA Cloning Vectors Bluescribe puc pJET pBR322

151
Plasmid features Size Plasmids range from about 1.0 kb for the smallest to over 250 kb for the largest plasmids copy number The factors that control copy number are not well understood. Some plasmids, especially the larger ones, are stringent and have a low copy number of perhaps just one or two per cell; others, called relaxed plasmids, are present in multiple copies of 50 or more per cell.

152
Viral vectors Delivery of genes by a virus is termed transduction and the infected cells are described as transduced.

153
Some other viral vectors use mostly in gene therapy & vaccination:
Lambda phage M13 bacteriophage Some other viral vectors use mostly in gene therapy & vaccination: Retroviruses (including lentiviruses) – integrating mammalian viruses; useful for stable/long-term gene expression Adeno-associated virus – replication-deficient parvoviruses, can integrate into a host cell genome; perspective vectors for gene Therapy Adenovirus – non-integrating DNA-genome virus; large application area from research to gene vaccination and anti-cancer Agents Poxvirus – large cytoplasmic DNA virus; perspective for development of gene vaccination
 – integrating mammalian. viruses; useful for stable/long-term gene expression. Adeno-associated virus – replication-deficient parvoviruses, can. integrate into a host cell genome; perspective vectors for gene. Therapy. Adenovirus – non-integrating DNA-genome virus; large. application area from research to gene vaccination and anti-cancer. Agents. Poxvirus – large cytoplasmic DNA virus; perspective for. development of gene vaccination.")
155
Lambda phage Lambda phage is a head & tail virus, containing double-stranded linear DNA as its genetic material, and a tail that can have tail fibers Usually, a "lytic cycle" ensues, where the lambda DNA is replicated many times and the genes for head, tail and lysis proteins are expressed. This leads to assembly of multiple new phage particles within the cell and subsequent cell lysis, releasing the cell contents, including virions that have been assembled, into the environment. However, under certain conditions the phage DNA may integrate itself into the host cell chromosome in the lysogenic pathway.

157
lytic cycle

158
Lambda Plaques

159
lysogenic pathway

162
M13 bacteriophage M13 is a filamentous bacteriophage composed of circular single stranded DNA (ssDNA) which is 6407 nucleotides long encapsulated in approximately 2700 copies of the major coat protein P8, and capped with 5 copies of two different minor coat proteins (P9, P6, P3) on the ends. The minor coat protein P3 attaches to the receptor at the tip of the F pilus of the host Escherichia coli. Infection with filamentous phages is not lethal; however, the infection causes turbid plaques in E. coli. It is a non-lytic virus. However a decrease in the rate of cell growth is seen in the infected cells. M13 plasmids are used for many recombinant DNA processes, and the virus has also been studied for its uses in nanostructures and nanotechnology.
 which is 6407 nucleotides long encapsulated in approximately 2700 copies of the major coat protein P8, and capped with 5 copies of two different minor coat proteins (P9, P6, P3) on the ends. The minor coat protein P3 attaches to the receptor at the tip of the F pilus of the host Escherichia coli. Infection with filamentous phages is not lethal; however, the infection causes turbid plaques in E. coli. It is a non-lytic virus. However a decrease in the rate of cell growth is seen in the infected cells. M13 plasmids are used for many recombinant DNA processes, and the virus has also been studied for its uses in nanostructures and nanotechnology.")
163
M13 Replication in E. coli Below are steps involved with replication of M13 in E. coli. Viral (+) strand DNA enters cytoplasm Complementary (-) strand is synthesized by bacterial enzymes DNA Gyrase, a type II topoisomerase, acts on double-stranded DNA and catalyzes formation of negative supercoils in double-stranded DNA Final product is parental replicative form (RF) DNA A phage protein, pII, nicks the (+) strand in the RF 3'-hydroxyl acts as a primer in the creation of new viral strand pII circulizes displaced viral (+) strand DNA Pool of progeny double-stranded RF molecules produced Negative strand of RF is template of transcription mRNAs are translated into the phage proteins Phage proteins in the cytoplasm are pII, pX, and pV, and they are part of the replication process of DNA. The other phage proteins are synthesized and inserted into the cytoplasmic or outer membranes. pV dimers bind newly synthesized single-stranded DNA and prevent conversion to RF DNA RF DNA synthesis continues and amount of pV reaches critical concentration DNA replication switches to synthesis of single-stranded (+) viral DNA pV-DNA structures from about 800 nm long and 8 nm in diamter pV-DNA complex is substrate in phage assembly reaction
 strand is synthesized by bacterial enzymes. DNA Gyrase, a type II topoisomerase, acts on double-stranded DNA and catalyzes formation of negative supercoils in double-stranded DNA. Final product is parental replicative form (RF) DNA. A phage protein, pII, nicks the (+) strand in the RF. 3 -hydroxyl acts as a primer in the creation of new viral strand. pII circulizes displaced viral (+) strand DNA. Pool of progeny double-stranded RF molecules produced. Negative strand of RF is template of transcription. mRNAs are translated into the phage proteins. Phage proteins in the cytoplasm are pII, pX, and pV, and they are part of the replication process of DNA. The other phage proteins are synthesized and inserted into the cytoplasmic or outer membranes. pV dimers bind newly synthesized single-stranded DNA and prevent conversion to RF DNA. RF DNA synthesis continues and amount of pV reaches critical concentration. DNA replication switches to synthesis of single-stranded (+) viral DNA. pV-DNA structures from about 800 nm long and 8 nm in diamter. pV-DNA complex is substrate in phage assembly reaction.")
165
cosmid A cosmid is a plasmid that contains a cos site from the lambda genome.

166
Cosmid vectors The vector replicates as a plasmid (it contains a ColE1 origin of replication), uses Ampr for positive selection and employs lambda phage packaging to select for recombinant plasmids carrying foreign DNA inserts 45 KB in size. Ligation of cosmid vector and foreign DNA fragments (SauIIIA partial digest fragments 45 kb in size) is similar to ligation into a lambda substitution vector. The desired ligation product is a concatemer of 45 kb foreign DNA fragment and 5 kb cosmid vector sequences.
, uses Ampr for positive selection and employs lambda phage packaging to select for recombinant plasmids carrying foreign DNA inserts 45 KB in size. Ligation of cosmid vector and foreign DNA fragments (SauIIIA partial digest fragments 45 kb in size) is similar to ligation into a lambda substitution vector. The desired ligation product is a concatemer of 45 kb foreign DNA fragment and 5 kb cosmid vector sequences.")
167
cosmid This concatemer is then packaged into viral particles (remember packaging is cos site to cos site) and these are used to infect E. coli where the cosmid vector replicates using the ColE1 orignin of replication. Phage packaging serves only to select for recombinant molecules and to transfer these long DNA molecues (50 kb total) into the bacterial host (50 kb fragments transform very inefficiently while phage infection is very efficient).
 and these are used to infect E. coli where the cosmid vector replicates using the ColE1 orignin of replication. Phage packaging serves only to select for recombinant molecules and to transfer these long DNA molecues (50 kb total) into the bacterial host (50 kb fragments transform very inefficiently while phage infection is very efficient).")
168
YAC yeast artificial chromosome
YAC vector is capable of carrying a large DNA fragment (up to 2 Mb), but its transformation efficiency is very low.
, but its transformation efficiency is very low.")
169
YAC Essential components of YAC vectors Procedure Centromers (CEN),
Telomeres (TEL) Autonomous replicating sequence (ARS) for proliferation in the host cell. Ampr for selective amplification Markers such as TRP1 and URA3 for identifying cells containing the YAC vector. Recognition sites of restriction enzymes (e.g., EcoRI and BamHI) Procedure 1- The target DNA is partially digested by EcoRI and the YAC vector is cleaved by EcoRI and BamHI. 2- Ligate the cleaved vector segments with a digested DNA fragment to form an artificial chromosome. 3- Transform yeast cells to make a large number of copies.
 Autonomous replicating sequence (ARS) for proliferation in the host cell. Ampr for selective amplification. Markers such as TRP1 and URA3 for identifying cells containing the YAC vector. Recognition sites of restriction enzymes (e.g., EcoRI and BamHI) Procedure. 1- The target DNA is partially digested by EcoRI and the YAC vector is cleaved by EcoRI and BamHI. 2- Ligate the cleaved vector segments with a digested DNA fragment to form an artificial chromosome. 3- Transform yeast cells to make a large number of copies.")
171
BAC Bacterial artificial chromosome
Bacterial Artificial Chromosomes (BAC) have been developed to hold much larger pieces of DNA than a plasmid can (They are able to hold up to 350 kb of DNA). BAC vectors were originally created from part of an F’ plasmid. In 1992, Hiroaki Shizuya took the parts of F’ that were important, cleaned it up, and turned it into a vector. With these vectors it is possible to study larger genes, several genes at once, or entire viral genomes.
 have been developed to hold much larger pieces of DNA than a plasmid can (They are able to hold up to 350 kb of DNA). BAC vectors were originally created from part of an F’ plasmid. In 1992, Hiroaki Shizuya took the parts of F’ that were important, cleaned it up, and turned it into a vector. With these vectors it is possible to study larger genes, several genes at once, or entire viral genomes.")
172
Common gene components
oriS, repE – F for plasmid replication and regulation of copy number. parA and parB for partitioning F plasmid DNA to daughter cells during division and ensures stable maintenance of the BAC. A selectable marker antibiotic resistance; some BACs also have lacZ at the cloning site for blue/white selection. T7 & Sp6 phage promoters for transcription of inserted genes.

173
BAC Bacterial artificial chromosome

174
Some of the scientific practices that have simplified with BAC vectors includes:
BACs are often used to sequence the genome of organisms in genome projects, for example the Human Genome Project. phylogenetic studies what the absolute minimum size of a genome could be. BAC vectors are also useful for studying pathogens, helpful in the development of vaccines. BAC vectors are playing a tremendous role in discovering new and powerful antibiotics in the environment. Discovering enzymes that are able to help clean up oil spills help farmers breed healthier farm animals process radioactive waste

176
Compare the capacity of vectors

177
Expression vector An expression vector, otherwise known as an expression construct, is generally a plasmid that is used to introduce a specific gene into a target cell. Once the expression vector is inside the cell, the protein that is encoded by the gene is produced by the cellular-transcription and translation machinery ribosomal complexes. The plasmid is frequently engineered to contain regulatory sequences that act as enhancer and promoter regions and lead to efficient transcription of the gene carried on the expression vector. The goal of a well-designed expression vector is the production of large amounts of stable messenger RNA, and therefore proteins. Expression vectors are basic tools for biotechnology and the production of proteins such as insulin that are important for medical treatments of specific diseases like diabetes.

178
Expression vector features
Promoter Shine-Dalgarno / kozak sequence Start codon Tags and fusion proteins Protease cleavage site Stop codon Transcription terminator Regulatory gene (repressor)
")
179
Expression vector

180
Regulatory gene (repressor)
Many promoters show leakiness in their expression i.e. gene products are expressed at low level before the addition of the inducer. This becomes a problem when the gene product is toxic for the host. This can be prevented by the constitutive expression of a repressor protein.

181
Promoter The promotor initiates transcription and is positioned nucleotides upstream of the ribosome binding site. The ideal promoter exhibits several desirable features: It is strong enough to allow product accumulation up to 50% of the total cellular protein. It has a low basal expression level (i.e. it is tightly regulated to prevent product toxicity). It is easy to induce.
. It is easy to induce.")
182
Shine-Dalgarno sequence
The Shine-Dalgarno (SD) sequence is required for translation initiation and is complementary to the 3'-end of the 16S ribosomal RNA. The efficiency of translation initiation at the start codon depends on the actual sequence. The concensus sequence is: 5'-TAAGGAGG-3'. It is positioned 4-14 nucleotides upstream the start codon with the optimal spacing being 8 nucleotides. The Kozak consensus sequence Kozak consensus or Kozak sequence, is a sequence which occurs on eukaryotic mRNA and has the consensus (gcc)gccRccAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another 'G‘. The Kozak consensus sequence plays a major role in the initiation of the translation process. The sequence was named after its discoverer, Marilyn Kozak.
 sequence is required for translation initiation and is complementary to the 3 -end of the 16S ribosomal RNA. The efficiency of translation initiation at the start codon depends on the actual sequence. The concensus sequence is: 5 -TAAGGAGG-3 . It is positioned 4-14 nucleotides upstream the start codon with the optimal spacing being 8 nucleotides. The Kozak consensus sequence. Kozak consensus or Kozak sequence, is a sequence which occurs on eukaryotic mRNA and has the consensus (gcc)gccRccAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another G‘. The Kozak consensus sequence plays a major role in the initiation of the translation process. The sequence was named after its discoverer, Marilyn Kozak.")
183
The Shine-Dalgarno sequence (or Shine-Dalgarno box), proposed by Australian scientists John Shine (b.1946) and Lynn Dalgarno (b.1935), Marilyn Kozak, PhD, professor of biochemistry at UMDNJ-Robert Wood Johnson Medical School. Lynn Dalgarno Marilyn Kozak John Shine

184
Tags and fusion proteins
N- or C-terminal fusions of heterologous proteins to short peptides (tags) or to other proteins (fusion partners) offer several potential advantages: Improved expression Fusion of the N-terminus of a heterologous protein to the C-terminus of a highly- expressed fusion partner often results in high level expression of the fusion protein. Improved solubility Fusion of the N-terminus of a heterologous protein to the C-terminus of a soluble fusion partner often improves the solubility of the fusion protein. Improved detection Fusion of a protein to either terminus of a short peptide (epitope tag) or protein which is recognized by an antibody or a binding protein (Western blot analysis) or by biophysical methods (e.g. GFP by fluorescence) allows for detection of a protein during expression and purification. Improved purification Simple purification schemes have been developed for proteins fused at either end to tags or proteins which bind specifically to affinity resins
 or to other proteins (fusion partners) offer several potential advantages: Improved expression. Fusion of the N-terminus of a heterologous protein to the C-terminus of a highly- expressed fusion partner often results in high level expression of the fusion protein. Improved solubility. Fusion of the N-terminus of a heterologous protein to the C-terminus of a soluble fusion partner often improves the solubility of the fusion protein. Improved detection. Fusion of a protein to either terminus of a short peptide (epitope tag) or protein which is recognized by an antibody or a binding protein (Western blot analysis) or by biophysical methods (e.g. GFP by fluorescence) allows for detection of a protein during expression and purification. Improved purification. Simple purification schemes have been developed for proteins fused at either end to tags or proteins which bind specifically to affinity resins.")
185
Protease cleavage site
Protease cleavage sites are often added to be able to remove a tag or fusion partner from the fusion protein after expression. However, cleavage is rarely complete and often additional purification steps are required. Start codon Initiation point of translation. In E. coli the most used start codon is ATG. GTG is used in 8% of the cases. TTG and TAA are hardly used. Stop codon Termination of translation. There are 3 possible stop codons but TAA is preferred because it is less prone to read-through than TAG and TGA. The efficiency of termination is increased by using 2 or 3 stop codons in series. Transcription terminator The transcription terminator reduces unwanted transcription and increases plasmid and mRNA stability.

186
pET-24(+) Vector
 Vector")
187
Use of linkers , adaptors & technique of homopolymer tailing
Can we made sticky end from blunt end DNA? Use of linkers , adaptors & technique of homopolymer tailing

188
Linkers: short pieces of double-stranded DNA
synthesized in the test tube It is blunt-ended contains a restriction site DNA ligase can attach linkers to the ends of target DNA molecules linkers, can be made in very large amounts and added into the ligation mixture at a high concentration. More than one linker will attach to each end of the DNA molecule

190
Adaptors: There is one potential drawback with the use of linkers.
Target DNA may containing same restriction endonuclease recognition sequence/ sequences. How we can avoid of this problem? Adaptors, Like linkers, are short synthetic oligonucleotides. But unlike linkers, an adaptor is synthesized so that it already has one sticky end.

191
Alkalin phosphatase

192
Ligation between inserted DNA & linear vector

193
technique of homopolymer tailing
A homopolymer is simply a polymer in which all the subunits are the same. Tailing involves using the enzyme terminal deoxynucleotidyl transferase to add a series of nucleotides onto the 3'-OH termini of a double stranded DNA molecule. If this reaction is carried out in the presence of just one deoxyribonucleotide a homopolymer tail is produced. In practice the poly( dG) and poly( dC) tails are not usually exactly the same length. Repair is therefore a two step process, using Klenow polymerase to fill in the nicks followed by DNA ligase to synthesize the final phosphodiester bonds.
 and poly( dC) tails are not usually exactly the same length. Repair is therefore a two step process, using Klenow polymerase to fill. in the nicks followed by DNA ligase to synthesize the final phosphodiester bonds.")
195
The repair steps

196
If the complementary homopolymer tails are longer than about 20 nucleotides, is often stable enough to be introduced into the host cell directly without repair . Once inside the host, the cell’s own DNA polymerase and DNA ligase repair the recombinant DNA molecule, completing the construction begun in the test tube.

197
T4 DNA Ligase Catalyzes the formation of a phosphodiester bond between juxtaposed 5' phosphate and 3' hydroxyl termini in duplex DNA or RNA. This enzyme will join blunt end and cohesive end termini as well as repair single stranded nicks in duplex DNA, RNA or DNA/RNA hybrids (1). Applications: Cloning of restriction fragments Joining linkers and adapters to blunt-ended DNA
. Applications: Cloning of restriction fragments. Joining linkers and adapters to blunt-ended DNA.")
198
P P 5’ 5’ OH OH OH OH 5’ P 5’ P P 5’ 5’ OH OH 5’ 5’ P 5’ OH OH 5’ P

199
P 5’ P 5’ OH OH OH OH 5’ P 5’ P P 5’ OH OH 5’ P P 5’ OH OH 5’ P

201
Alkaline Phosphatase Alkaline Phosphatase catalyzes the removal of 5´ phosphate groups from DNA, RNA, ribo- and deoxyribonucleoside triphosphates. Since CIP-treated fragments lack the 5´ phosphoryl termini required by ligases, they cannot self-ligate (1). This property can be used to decrease the vector background in cloning strategies. Source: Calf intestinal mucosa (CIP) or Shrimp (SAP) Applications: Removing 5´ phosphates from DNA, RNA, rNTPs and dNTPs Preparation of templates for 5´end labeling Prevention of recircularization of cloning vectors Dephosphorylation of serine, threonine and tyrosine residues in protein
. This property can be used to decrease the vector background in cloning strategies. Source: Calf intestinal mucosa (CIP) or Shrimp (SAP) Applications: Removing 5´ phosphates from DNA, RNA, rNTPs and dNTPs. Preparation of templates for 5´end labeling. Prevention of recircularization of cloning vectors. Dephosphorylation of serine, threonine and tyrosine residues in protein.")
202
Construct of DNA & cDNA library

203
Types of libraries The DNA cloned in libraries depends on the purposes of the investigators Genomic library: A collection of fragments representing all DNA in the chromosomes of a cell. Genomic libraries are made from total nuclear DNA of an organism. Chromosome-specific libraries: are made from the DNA of purified isolated chromosomes. cDNA library: Collection of DNAs representing the genes expressed in cell or tissue type. cDNA libraries are made from the mRNA populations of particular tissues. There are thus liver, heart, leaf, root, etc. cDNA libraries. The advantage of cDNA library is that it contains only the coding region of a genome cDNA expression libraries: A collection of expressed genes cloned into a vector next to a regulatory region so that the protein will be produced under conditions controlled by the experimenter. For example, DNA can be cloned into a vector to create a fusion protein used to test a gene product for interaction with other proteins. Electronic libraries: With the human genome project, the DNA sequence from several model organisms as well as humans is being determined. This information serves as a virtual DNA library that can be analyzed via computer

205
Genomic Library Genomic DNA Randomly cut Ligate Transform

206
DIGESTION OF GENOMIC DNA
Complete digestion with an endonuclease will result in a library containing no overlapping fragments: Complete digestion has these restriction: 1- produce many of fragment that construction of library and screening is difficult 2- If a sequence has more than 1 recognision site for an enzyme, caused cloning of fragment in form of 2 or more unoverlapping fragments. In this condition, we need walking clones to findout overlapping fragments 3- Because of nonrandomly disribution of restriction sites on DNA, such a sequence may located on a fragment which so very large or small that can’t be cloned. Solving of problems: 1- mechanical shearing: hasn’t desirable efficiency 2- partial digestion: main method

207
PARTIAL DIGESTION Adventages:
Incomplete Digestion of Genomic DNA will allow identification of sequence overlaps However, incomplete digestion will result in a library containing overlapping fragments: Use the reduction of enzyme concentration or time of activity Adventages: Thus, the sequence information obtained from one clone will allow the isolation of clones containing neighboring (overlapping) sequence information. This can allow large contiguous stretches of sequence information to be obtained ("Chromosome Walking"). overlapping fragments allows larger physical maps to be constructed as contiguous chromosomal regions (contigs) are put together from the sequence data Reduction the size of library (large size of fragment): number of clones needed to fully represent the genome Without information abuot the location of restriction sites near the tag sequence , it can be isolate the fragments
 sequence information. This can allow large contiguous stretches of sequence information to be obtained ( Chromosome Walking ). overlapping fragments allows larger physical maps to be constructed as contiguous chromosomal regions (contigs) are put together from the sequence data. Reduction the size of library (large size of fragment): number of clones needed to fully represent the genome. Without information abuot the location of restriction sites near the tag sequence , it can be isolate the fragments.")
208
a “contig” Partial restriction enzyme digestion
allows cloning of overlapping fragments a “contig” isolation of ~20 kb fragments provides optimally sized DNAs for cloning in bacteriophage partial digestion with a frequent-cutter (4-base cutter) allows production of overlapping fragments, since not every site is cut overlapping fragments insures that all sequences in the genome are cloned
 allows production. of overlapping fragments, since not every site is cut. overlapping fragments insures that all sequences in the genome are cloned.")
209
All possible sites: Results of a partial digestion: = uncut = cut

210
Restriction endonucleases
Restriction enzymes cut DNA into specific fragments Frequency of cutting (if frequency of all bases are equall) 4-base cutter 44 = bp 5-base cutter 45 = 1,024 bp 6-base cutter 46 = 4,096 bp 8-base cutter 48 = 65,536 bp In practice, the actual number of fragments is quite different because the distribution of nucleotides is non-random and most organismes do not have an equal number of the four bases for example; human DNA has overall only 40% G + C and the frequency of the dinucleotide CpG is only 20% of that expected. In addition many cytosine residues are methylated and this can prevent restriction endonucleases digetion. E.g.: Not I (8-base cutter 48 = 65,536 bp or 65.5 Kb) but produce fragments with average size of Kb
 4-base cutter 44 = 256 bp. 5-base cutter 45 = 1,024 bp. 6-base cutter 46 = 4,096 bp. 8-base cutter 48 = 65,536 bp. In practice, the actual number of fragments is quite different because the distribution. of nucleotides is non-random and most organismes do not have an equal number of. the four bases. for example; human DNA has overall only 40% G + C and the frequency of the dinucleotide CpG is only 20% of that expected. In addition many cytosine residues are methylated. and this can prevent restriction endonucleases digetion. E.g.: Not I (8-base cutter 48 = 65,536 bp or 65.5 Kb) but produce fragments. with average size of Kb.")
211
cuts DNA into 256 bp average-sized fragments in a random sequence
4-base cutter: cuts DNA into 256 bp average-sized fragments in a random sequence every 256 bp: NO 256 bp average-size fragments: YES Bar = 256 b

212
Construct of cDNA library
Procedure: 1- Total RNA extraction (tRNA, rRNA, mRNA, snRNA,… ) 2- Isolation of mRNA from Total RNA 3- Synthesis of sscDNA from mRNA 4- produce of dscDNA from mRNA 5-Insertion of dscDNA in to the cloning vectors 6- Transformation (transduction) in to the host cells
 2- Isolation of mRNA from Total RNA 3- Synthesis of sscDNA from mRNA 4- produce of dscDNA from mRNA 5-Insertion of dscDNA in to the cloning vectors 6- Transformation (transduction) in to the host cells")
213
Isolation of mRNA from Total RNA

214
ExactSTART™ Full-Length cDNA Library Cloning Kit

215
cDNA library construction: Replacement synthesis of second strand of cDNA
5’ AAAAA 3’ mRNA (all mRNAs in cell) anneal oligo(dT) primers of bases in length 5’ AAAAA TTTTT 3’ 5’ 3’ add reverse transcriptase and dNTPs 5’ 3’ AAAAA TTTTT 3’ 5’ cDNA add RNaseH (specific for the RNA strand of an RNA-DNA hybrid) and carry out a partial digestion 5’ 3’ AA TTTTT short RNA fragments serve as primers for second strand synthesis using DNA polymerase I
 anneal oligo(dT) primers of bases in length. 5’ AAAAA. TTTTT. 3’ 5’ 3’ add reverse transcriptase and dNTPs. 5’ 3’ AAAAA. TTTTT. 3’ 5’ cDNA. add RNaseH (specific for the RNA strand of an RNA-DNA. hybrid) and carry out a partial digestion. 5’ 3’ AA. TTTTT. short RNA fragments serve as primers for. second strand synthesis using DNA polymerase I.")
216
5’ 3’ AAAAA TTTTT AAA TTTTT 5’ 3’ AAA TTTTT 5’ 3’ 5’ 3’ AAAAA TTTTT
short RNA fragments serve as primers for second strand synthesis using DNA polymerase I AAA TTTTT 5’ 3’ DNA polymerase I removes the remaining RNA with its 5’ to 3’ exonuclease activity and continues synthesis AAA TTTTT 5’ 3’ DNA ligase seals the gaps 5’ 3’ AAAAA TTTTT double-stranded cDNA

217
5’ 3’ AAAAA TTTTT AAAAANNNNNNNNG TTTTTNNNNNNNNCTTAA 5’ 3’
EcoRI linkers are ligated to both ends using DNA ligase AAAAANNNNNNNNG TTTTTNNNNNNNNCTTAA 5’ 3’ AATTCNNNNNNNN GNNNNNNNN double-stranded cDNA copies of mRNA with EcoRI cohesive ends are now ready to ligate into a bacteriophage lambda vector cut with EcoRI

219
primed synthesised of second strand of cDNA

227
Target DNA Desirable fragment

228
Cloning process Source DNA Cloning vector Target DNA Enzymatically
linearize Amplified by PCR & then digestion Enzymatically fragmented Ligation Transformation in to the host cell Amplification

229
Gene transfer methods

230
Transient vs. Stable Transient transfection
In transient transfection, the transfected DNA is not integrated into host chromosome. DNA is transferred into a recipient cell in order to obtain a temporary but high level of expression of the target gene. Stable transfection Stable transfection is also called permanent transfection. By the stable transfection, the transferredDNA is integrated (inserted) into chromosomal DNA and the genetics of recipient cells is permanent changed.
 into chromosomal DNA and the genetics of recipient cells is permanent changed.")
231
Transfection Methods Generally, there are 9 ways for gene transfer:
(1) Lipid-mediated method; (2) Calcium-phosphate mediated; (3) DEAE-dextran-mediated; (4) Electroporation; (5) Biolistics; (6) Viral vectors; (7) Polybrene; (8) Laser transfection; (9) Gene transfection enhanced by elevated temperature (Sambrook, 2001).
 Lipid-mediated method; (2) Calcium-phosphate mediated; (3) DEAE-dextran-mediated; (4) Electroporation; (5) Biolistics; (6) Viral vectors; (7) Polybrene; (8) Laser transfection; (9) Gene transfection enhanced by elevated temperature (Sambrook, 2001).")
232
Indirect gene transfer (Vector-mediated)
Direct gene transfer (Vectorless)
")
233
Direct gene transfer (Vectorless)
Chemical methods Electroporation Biolistic, Particle gun delivery Lipofection Microinjection and macroinjection Laser induced Fibremediated

234
Chemical mediated gene transfer
Chemicals like polyethylene glycol (PEG) and dextran sulphate induce DNA uptake into plant protoplasts. Calcium phosphate is also used to transfer DNA into cultured cells.
 and dextran sulphate induce DNA uptake into plant protoplasts. Calcium phosphate is also used to transfer DNA into cultured cells.")
235
Microinjection where the DNA is directly injected into protoplasts or cells (specifically into the nucleus or cytoplasm) using fine tipped ( micrometer diameter) glass needle or micropipette. This method of gene transfer is used to introduce DNA into large cells such as oocytes, eggs, and the cells of early embryo.
 using fine tipped ( micrometer diameter) glass needle or micropipette. This method of gene transfer is used to introduce DNA into large cells such as oocytes, eggs, and the cells of early embryo.")
236
Electroporation involves a pulse of high voltage applied to protoplasts/cells/ tissues to make transient (temporary) pores in the plasma membrane which facilitates the uptake of foreign DNA. The cells are placed in a solution containing DNA and subjected to electrical shocks to cause holes in the membranes. The foreign DNA fragments enter through the holes into the cytoplasm and then to nucleus.
 pores in the plasma membrane which facilitates the uptake of foreign DNA. The cells are placed in a solution containing DNA and subjected to electrical shocks to cause holes in the membranes. The foreign DNA fragments enter through the holes into the cytoplasm and then to nucleus.")
239
Lipofection

240
liposomes A vesicle is a small bubble enclosed by lipid bilayer. Vesicles can form naturally, for example, during the endocytosis, or they may be prepared. Artificially prepared vesicles are known as liposomes.

241
Lipofection Lipofection (or liposome transfection) is a technique used to inject genetic material into a cell by means of liposomes, which are vesicles that can easily merge with the cell membrane since they are both made of a phospholipid bilayer. Lipofection generally uses a positively charged (cationic) lipid to form an aggregate with the negatively charged (anionic) genetic material. A net positive charge on this aggregrate has been assumed to increase the effectiveness of transfection through the negatively charged phospholipid bilayer.
 is a technique used to inject genetic material into a cell by means of liposomes, which are vesicles that can easily merge with the cell membrane since they are both made of a phospholipid bilayer. Lipofection generally uses a positively charged (cationic) lipid to form an aggregate with the negatively charged (anionic) genetic material. A net positive charge on this aggregrate has been assumed to increase the effectiveness of transfection through the negatively charged phospholipid bilayer.")
242
Lipofection advantages
high efficiency ability to transfect all types of nucleic acids in a wide range of cell types ease of use reproducibility low toxicity In addition, this method is suitable for all transfection applications (transient, stable, co-transfection, reverse, sequential or multiple transfections…). High throughput screening assay and has also shown good efficiency in some in vivo models.
. High throughput screening assay and has also shown good efficiency in some in vivo models.")
243
biolistic

245
biolistic

246
Gene gun

247
Gene transfer methods Vectorless or direct gene transfer
Chemical mediated gene transfer(polyethylene glycol(PEG)) Microinjection / Macroinjection Electroporation Particle gun/Particle bombardment (gene gun) Liposome mediated gene transfer or Lipofection Vector-mediated or indirect gene transfer Agrobacterium as gene transfer system Viral mediated gene transfer
) Microinjection / Macroinjection. Electroporation. Particle gun/Particle bombardment (gene gun) Liposome mediated gene transfer or Lipofection. Vector-mediated or indirect gene transfer. Agrobacterium as gene transfer system. Viral mediated gene transfer.")
248
DIRECT GENE TRANSFER Chemical methods Electroporation Particle gun
Lipofection Microinjection And macroinjection Pollen transformation Delivery Via growing pollen tubes Laser induced Fiber mediated

249
Agrobacterium tumefaciens
Agrobacterium tumefaciens (updated scientific name: Rhizobium radiobacter) is the causal agent of crown gall disease (the formation of tumours) in over 140 species of dicot. It is a rod shaped, Gram negative soil bacterium. A. tumefaciens is an alphaproteobacterium of the family Rhizobiaceae, which includes the nitrogen fixing legume symbionts. Unlike the nitrogen fixing symbionts, tumor producing Agrobacterium are pathogenic and do not benefit the plant.
 is the causal agent of crown gall disease (the formation of tumours) in over 140 species of dicot. It is a rod shaped, Gram negative soil bacterium. A. tumefaciens is an alphaproteobacterium of the family Rhizobiaceae, which includes the nitrogen fixing legume symbionts. Unlike the nitrogen fixing symbionts, tumor producing Agrobacterium are pathogenic and do not benefit the plant.")
250
Tumour-inducing plasmid (Ti plasmid(
a tumour-inducing plasmid (Ti plasmid or pTi), of 200 kb, which contains the T-DNA and all the genes necessary to transfer it to the plant cell. ( Many strains of A. tumefaciens do not contain a pTi.)
, of 200 kb, which contains the T-DNA and all the genes necessary to transfer it to the plant cell. ( Many strains of A. tumefaciens do not contain a pTi.)")
252
Transfer of T-DNA into the plant cell
A: Agrobacterium tumefaciens B: Agrobacterium genome C: Ti Plasmid : a: T-DNA , b: Vir genes , c: Replication origin , d: Opines catabolism genes D: Plant cell E: Mitochondria F: Chloroplast G: Nucleus

253
RB LB Disarm RB LB promoter terminator

254
Ti plasmid binary vector Large plasmid Small plasmid Helper Ti plasmid
(pBI121) Virulence genes(Vir genes), ori T-DNA (nptII, target gene), Ecoli ori, Agro ori, nptIII
 Virulence genes(Vir genes), ori. T-DNA (nptII, target gene), Ecoli ori, Agro ori, nptIII.")
255
pBI121 is a Shuttle vector

256
Binary vector

258
Agrobacterium rhizogenes
Agrobacterium rhizogenes (updated scientific name: Rhizobium rhizogenes) is a Gram negative soil bacterium that produces hairy root disease in dicotyledonous plants. Containing root inducing plasmid (Ri plasmid)
 is a Gram negative soil bacterium that produces hairy root disease in dicotyledonous plants. Containing root inducing plasmid (Ri plasmid)")
Similar presentations








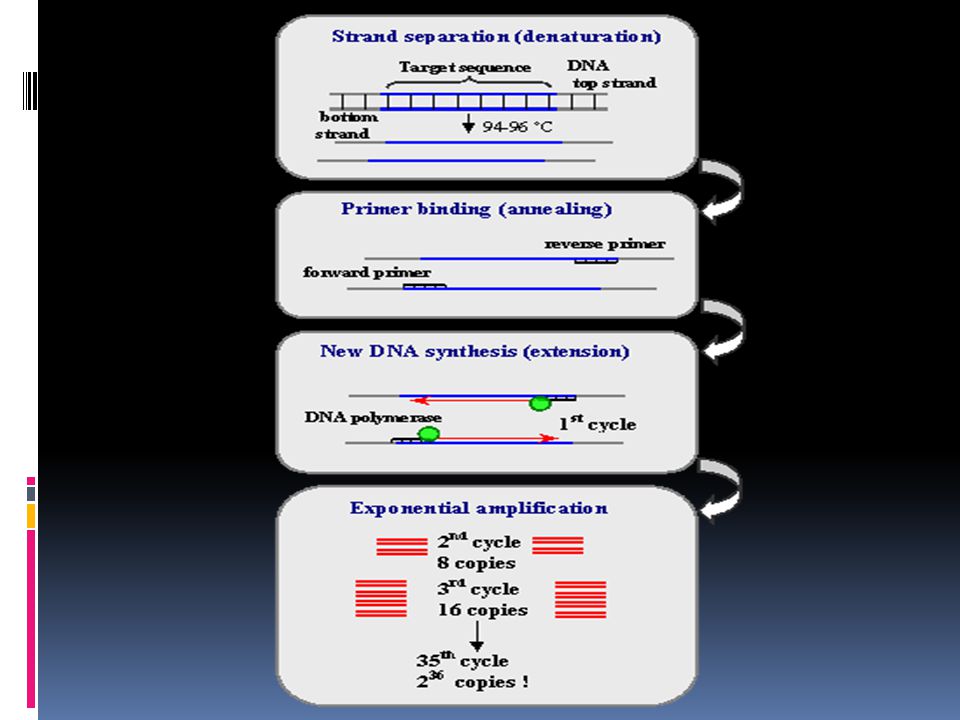




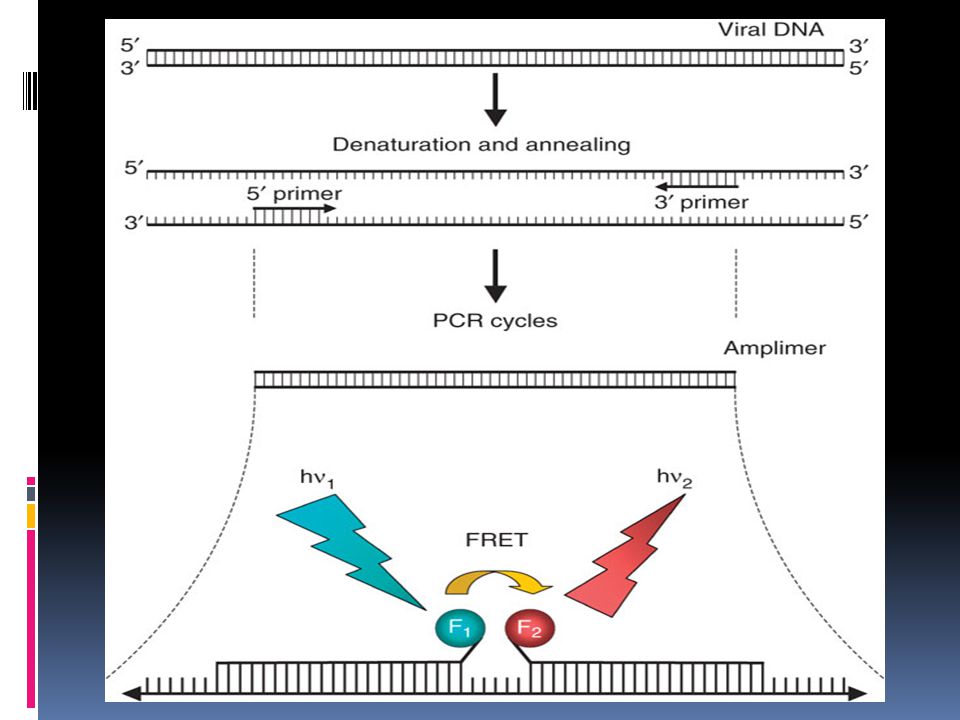

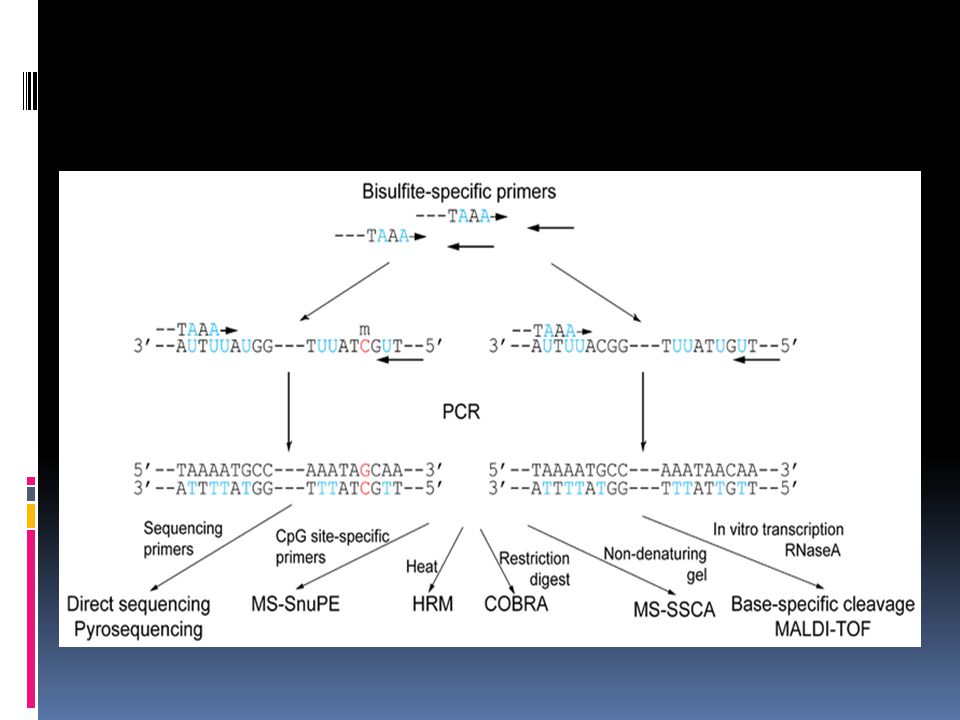





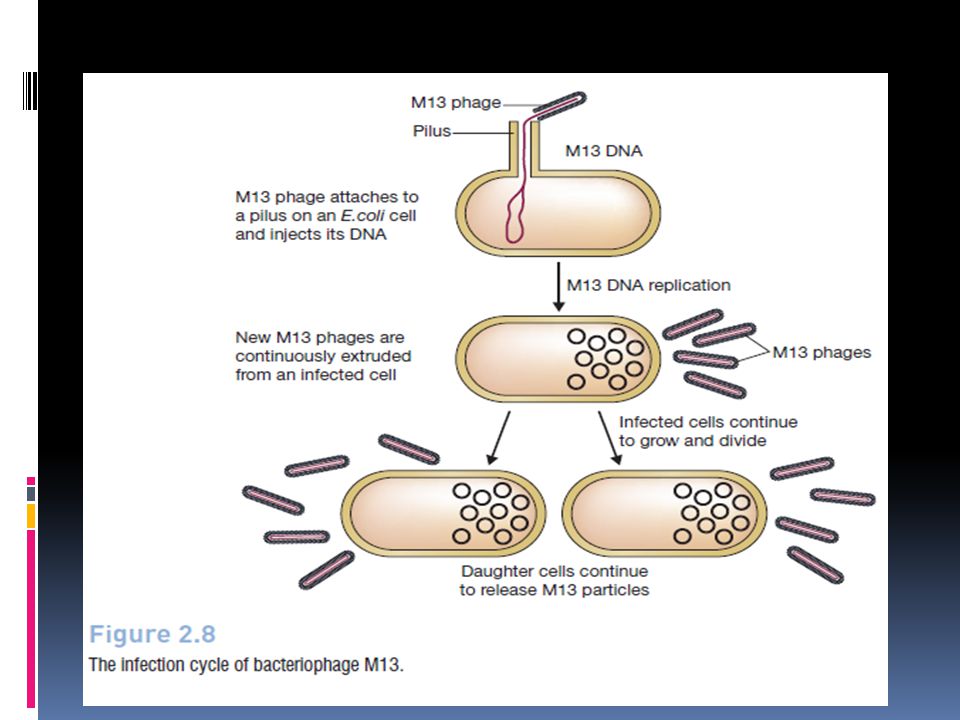

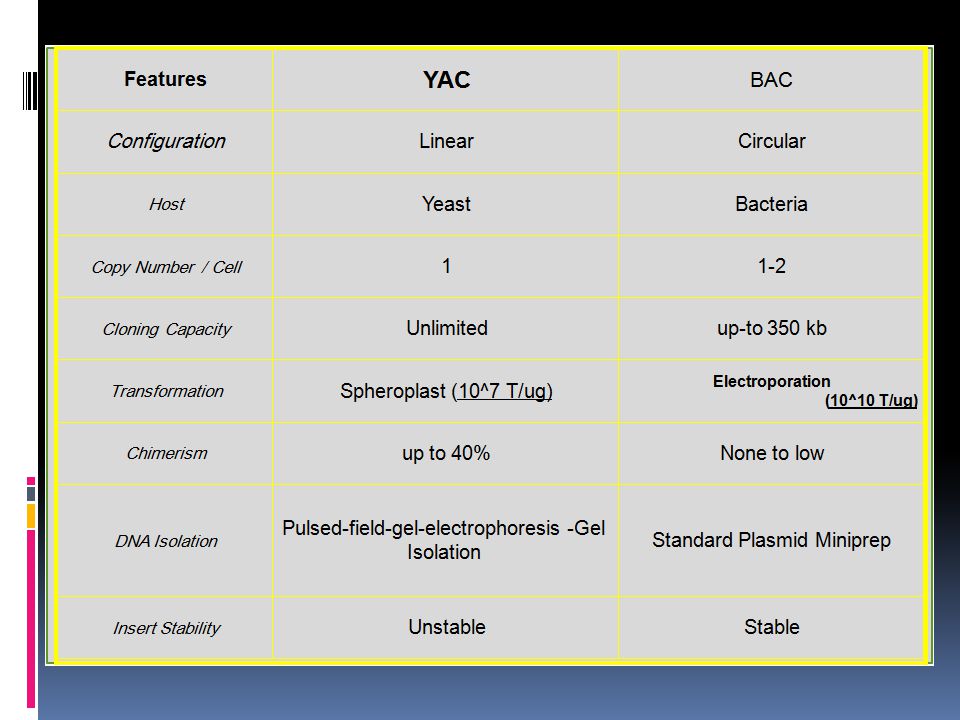




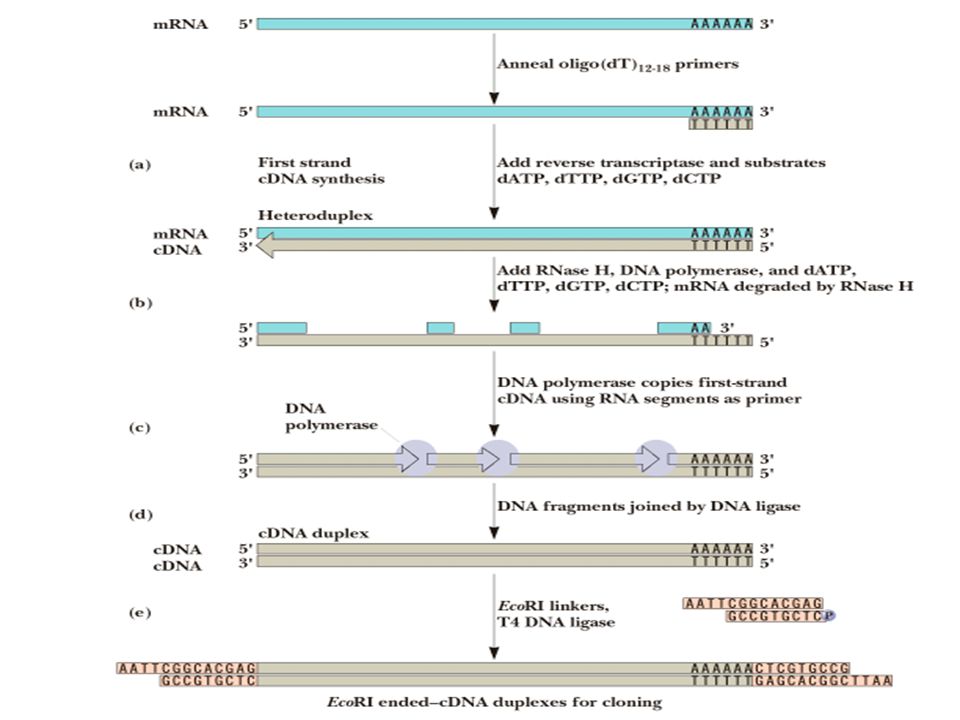
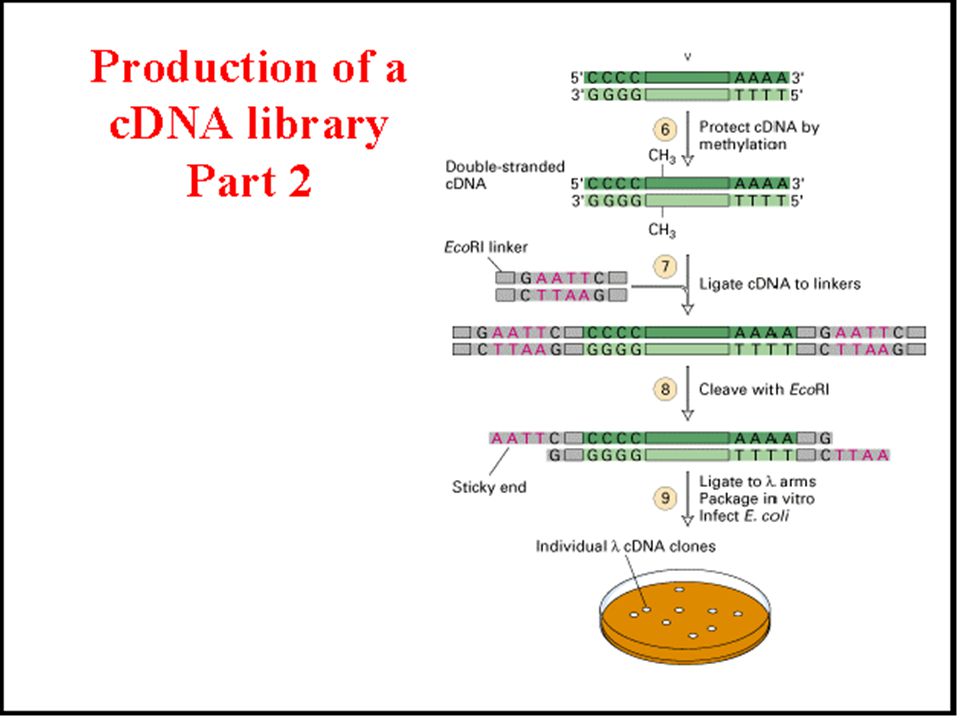








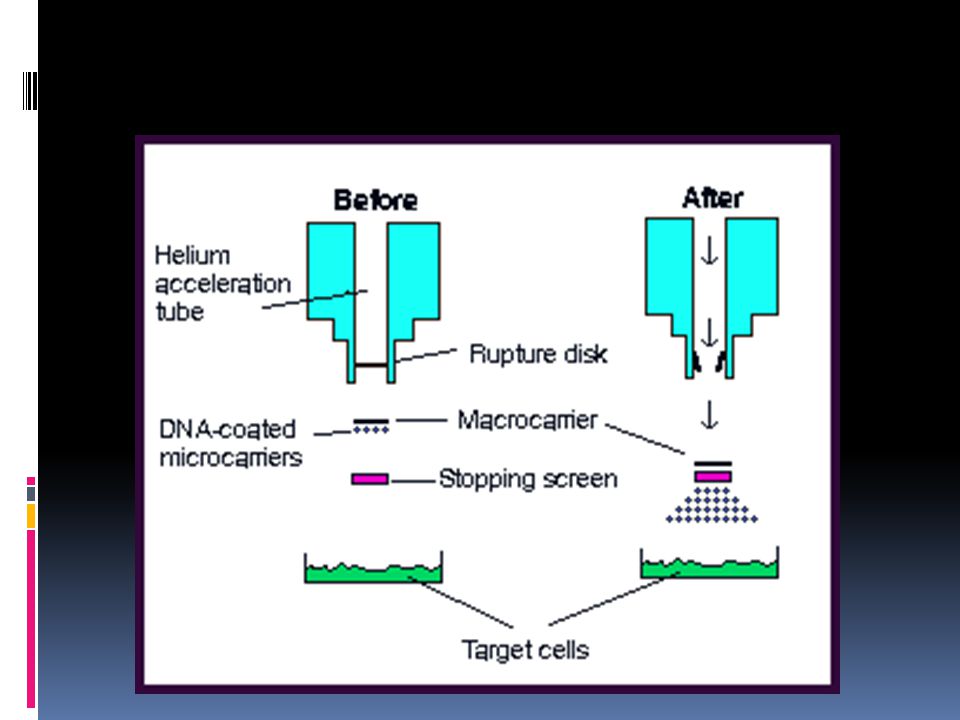
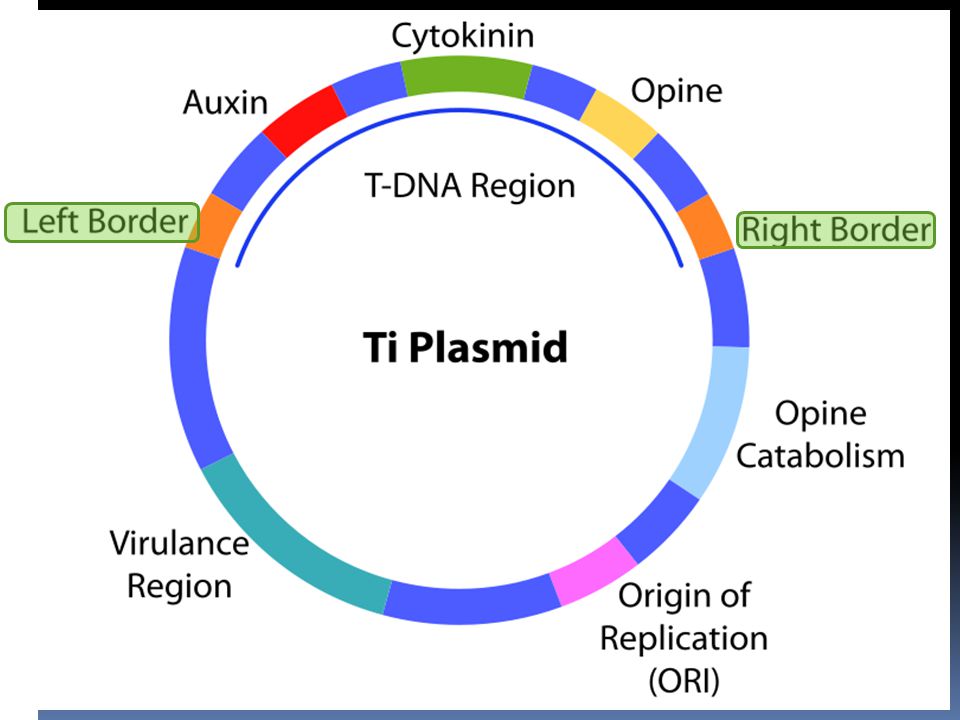












 Analysis of DNA (Sequencing) Chemical Synthesis.>")


 Analysis of DNA (Sequencing) Chemical Synthesis of DNA.>")

 Lecture 11 Biotechnology (Text Chapters: 10.15-10.17; 31.1-31.10)>")

