Download presentation
Presentation is loading. Please wait.

1
Amyotropic Lateral Sclerosis (ALS)
Jerry Carley RN, MSN, MA, CNE AH II Summer 2010

2
Concept Map: Selected Topics in Neurological Nursing
PATHOPHYSIOLOGY Traumatic Brain Injury Spinal Cord Injury Specific Disease Entities: Amyotropic Lateral Sclerosis Multiple Sclerosis Huntington’s Disease Alzheimer’s Disease Myasthenia Gravis Guillian-Barre’ Syndrome Meningitis Parkinson’s Disease ASSESSMENT Physical Assessment Inspection Palpation Percussion Auscultation ICP Monitoring “Neuro Checks” Lab Monitoring PHARMACOLOGY --Decrease ICP --Disease / Condition Specific Meds Care Planning Plan for client adl’s, Monitoring, med admin., Patient education, more…based On Nursing Process: A_D_P_I_E Nursing Interventions & Evaluation Execute the care plan, evaluate for Efficacy, revise as necessary

3
ALS is also known as Lou Gehrig's Disease, after the famous baseball player who died of the disease in 1941

4
Characteristics of ALS
Disease of the motor system Progressive muscle atrophy Fatal (2 – 5 years) d/t respiratory failure
 d/t respiratory failure.")
5
Characteristics Most commonly diagnosed in s Affects men more often than women The disease has no racial, socioeconomic, or ethnic boundaries

6
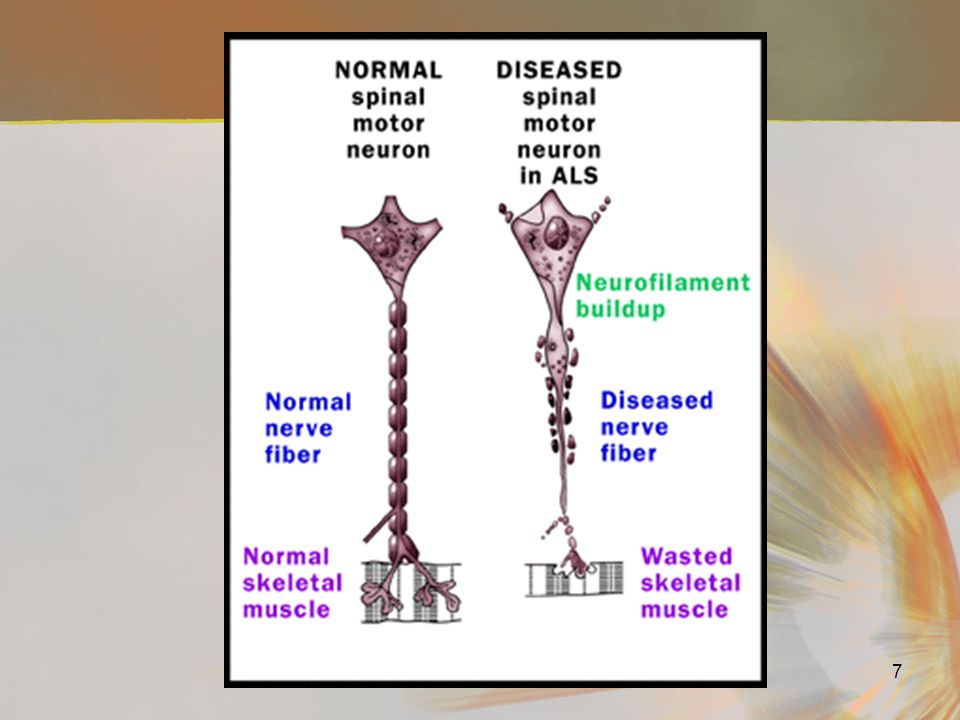
What’s in a Name ? Amyotrophy refers to the atrophy (progressive muscle wasting) Lateral sclerosis refers to demyelination followed by hardening of the spinal column from buildup of scar tissue (sclerosis = scar) As the disease progresses, it will move up the affected leg or arm until eventually all muscle groups become involved. This spread into all muscle groups is the defining characteristic of ALS
 As the disease progresses, it will move up the affected leg or arm until eventually all muscle groups become involved. This spread into all muscle groups is the defining characteristic of ALS.")
8
Why ? ... Etiology….. Mitochondrial dysfunction?
Genetic defect (chromosome 21) suggests the disease is inherited and accounts for 5 to 10% Environmental factors, since the disease tends to cluster in geographical pockets? (Extremely high incidence of ALS has been observed in Guam and the Trust Territories of the Pacific ) Free radical damage? Glutamate excitotoxicity? (Apoptosis / Programmed Cell Death)
 suggests the disease is inherited and accounts for 5 to 10% Environmental factors, since the disease tends to cluster in geographical pockets (Extremely high incidence of ALS has been observed in Guam and the Trust Territories of the Pacific ) Free radical damage Glutamate excitotoxicity (Apoptosis / Programmed Cell Death)")
9
Signs and Symptoms Difficulty swallowing (dyphagia)
Slurred speech (dysarthria) Fatigue Fasciculations of tongue (twitching) while at rest Usually the first muscles affected are those in the arms and legs (Walking or climbing stairs may be difficult, may drop things, fall, experience muscle cramps. The arms and legs may feel especially tired. If the hands are affected, may have difficulty picking up small objects or turning keys) Overview Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig's disease, is a progressive neuromuscular disease that weakens and eventually destroys motor neurons (components of the nervous system that connect the brain with the skeletal muscles). Skeletal muscles are involved with voluntary movements, such as walking and talking. The motor neurons transmit the command to move from the brain to the skeletal muscles, which respond by contracting. A person with ALS usually presents with problems in dexterity or gait resulting from muscle weakness, or with difficulty speaking or swallowing. Sphincter control, sensory function, intellectual ability, and skin integrity are preserved. Patients become paralyzed and often require ventilation and surgery to provide a new opening in the stomach (gastrostomy). Loss of respiratory function is ultimately the cause of death. Incidence and Prevalence Approximately 30,000 patients in the United States currently have ALS. The disease has no racial, socioeconomic, or ethnic boundaries. The life expectancy of ALS patients is usually 3 to 5 years after diagnosis. ALS is most commonly diagnosed in middle age and affects men more often than women. Risk Factors Risk factors include an inherited genetic defect, which accounts for 5–10% of cases of familial ALS (FALS) in the United States. FALS is linked to a genetic defect on chromosome 21. This gene codes for an enzyme called superoxide dismutase (SOD), an antioxidant that protects motor neurons from free radical damage (i.e., molecules introduced to the body, or produced by body processes that interact and cause cellular damage). More than 60 different mutations that cause SOD to lose its antioxidant properties have been found. However, only 40% of familial ALS cases are linked to SOD mutations, so there may be other unknown genetic defects involved. In the United States, 90–95% of ALS cases are sporadic. Sporadic ALS appears to be increasing worldwide. The causes are not clear, yet some evidence suggests that the immune system may be involved. Excessive levels of glutamate can overstimulate motor neurons and cause them to die. Glutamate is one of the most important neurotransmitters for healthy brain function. Neurotransmitters are chemicals that transmit signals from one nerve to another. In Guamanian ALS, a dietary neurotoxin is the risk factor. The suspected neurotoxin is an amino acid (BMAA) found in the seed of the cycad Cyas cirinalis, a tropical plant found in Guam, which was used to make flour and was a major dietary component during the 1950s and the early 1960s, when this type of ALS had an exceptionally high incidence. Causes The cause of ALS is not completely understood. Researchers and physicians suspect viruses, neurotoxins (especially in Guamanian ALS), heavy metals, DNA defects (especially in familial ALS), immune system abnormalities, and enzyme abnormalities. Signs and Symptoms Once a motor neuron degenerates completely, the muscle that it controls no longer receives impulses from the brain. Approximately 60% of ALS patients experience muscle weakness and stiffness as the initial symptom. Usually the first muscles affected are those in the arms and legs. Walking or climbing stairs may be difficult. The patient may drop things, fall, experience muscle cramps, and laugh or cry uncontrollably. The arms and legs may feel especially tired. If the hands are affected, the patient may have difficulty picking up small objects or turning keys. Speech problems, such as slurring, hoarseness, or decreased volume may also occur. Signs and symptoms include the following: Absence of spinal reflexes (areflexia) Loss of muscle tone (hypotonia) Muscle twitching (fasciculations) Muscle wasting (atrophy) The signs that the upper motor neurons are affected include the following: Excessive salivation Extension of the big toe and abduction of the rest of the toes in response to lightly stroking the sole of the foot (Babinski's sign) Reduced muscle tone, or hypotonia, and rigidity (spasticity) Hyperactive tendon reflexes Impaired speech (dysarthria) Impaired swallowing (dysphagia) Rapidly alternating muscle contractions and relaxations (clonus) As symptoms progressively worsen, the patient's muscles atrophy causing spasticity, stiffness, abnormal movements, and alterations in gait and manual dexterity. Twitching may occur in the tongue and in affected limbs. The patient may experience muscle pain and muscle cramps. Some patients experience more difficulty swallowing saliva and liquids than solid food. Excessive salivation and difficulty swallowing may cause drooling. When respiratory muscles weaken, the patient may require a ventilator. ALS patients often experience fear, anxiety, and depression. Overview Diagnosis ALS is difficult to diagnose because the symptoms are similar to those of other neuromuscular disorders, many of which are treatable. The diagnosis is usually based on a complete neurological examination and clinical tests. The neurological exam usually shows evidence of muscle weakness (localized or widespread, depending on the extent of the disease). The exam also reveals muscle atrophy. The muscles may be so stiff that when the neurologist moves them, they continue to move abnormally afterward. When the neurologist tests the "knee jerk" reaction, the movement is abnormally quick. Because ALS affects the skeletal, voluntary muscles, the neurological exam does not reveal abnormalities in the sensory reflexes (i.e., vision, hearing, taste, smell, touch, or bowel and bladder control). Tests Nerve conduction velocity (NCV) and electromyography (EMG) help diagnose nerve and muscle disorders. These tests, which can be used in combination, are often referred to as EMG/NCV studies. NCV is administered before EMG and measures the speed at which nerves transmit electrical signals. During NCV, electrodes are placed on the skin over a nerve that supplies a specific muscle or muscle group. A mild, brief electrical stimulus is delivered through the electrode and the response of the muscle is detected, amplified, and displayed. The strength of the signal is also measured. Neurological conditions can cause the NCV to slow down or to be slower on one side of the body. The strength of the response also provides clues to help with diagnosis and to determine the extent of the disease. EMG measures nerve impulses within the muscles. Tiny electrodes are placed in the muscles in the arms and legs and the electronic responses are observed using an instrument that displays movement of an electric current (oscilloscope). As muscles contract, they emit a weak electrical signal that can be detected, amplified, and tracked, providing information about how well the muscles are working. These responses are abnormal in cases of ALS. Tests may be performed to rule out other neurological disorders. Magnetic resonance imaging (MRI scan) may be used to rule out spinal cord disease. Blood tests may be done to detect the presence of heavy metals such as lead in the blood. Laboratory tests may detect abnormal proteins or hormone levels associated with other neurological diseases. A lumbar puncture or spinal tap may be performed to analyze the cerebrospinal fluid for genetic abnormalities (e.g., viral, autoimmune, neurotoxic). Overview Treatment There is no cure for ALS. Treatment focuses on relieving symptoms and maintaining an optimal quality of life. Treatment is based on individual therapy and the continual adaptation of medications. Riluzole (Rilutek®) is one of the few drugs effective against ALS and may prevent progression and prolong life for a few months or so. Riluzole decreases the release of glutamate. Side effects include the following: Dizziness Elevated liver enzymes Reduced leukocytes in the blood (granulocytopenia) Weakness (asthenia) Baclofen (Lioresol®) or tizanadine (Zanaflex®) may relieve spasticity. Side effects include increased weakness, sedation, and dizziness. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen or naproxen may relieve general discomfort. Due to potentially severe gastrointestinal and cardiovascular side effects, NSAIDs should only be used as instructed. Tramadol (Ultram®) is often prescribed for pain relief. Physical therapy is an important part of treatment and helps to relieve cramping and muscular pain. Passive stretching helps to avoid permanent contraction of muscles (contractures) that may cause joint problems. Other therapies, such as occupational therapy and speech therapy, are also used in treatment. ALS patients require a diet of high-energy foods that are easy to swallow. Patients may benefit from a nutritionist. If the patient is not able to maintain adequate nutrition, a percutaneous endoscopic gastrostomy (PEG), or feeding tube, is usually inserted. Some ALS patients may also need pulmonary consultants and respiratory therapists to assist breathing. Fewer than 5% of patients use long-term ventilation support. Depression is very common among ALS patients. Antidepressant medication and counseling can help patients and their families cope. Prognosis ALS is a terminal illness. Fifty percent of patients die within 3 years of diagnosis; 20% live 5 years; and 10% live 10 years. Hospice care can provide comfort and dignity to patients and their loved ones. Resources for patients and caregivers are available from the ALS Association and the ALS Division of t
 Fatigue. Fasciculations of tongue (twitching) while at rest. Usually the first muscles affected are those in the arms and legs. (Walking or climbing stairs may be difficult, may drop things, fall, experience muscle cramps. The arms and legs may feel especially tired. If the hands are affected, may have difficulty picking up small objects or turning keys) Overview Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig s disease, is a progressive neuromuscular disease that weakens and eventually destroys motor neurons (components of the nervous system that connect the brain with the skeletal muscles). Skeletal muscles are involved with voluntary movements, such as walking and talking. The motor neurons transmit the command to move from the brain to the skeletal muscles, which respond by contracting. A person with ALS usually presents with problems in dexterity or gait resulting from muscle weakness, or with difficulty speaking or swallowing. Sphincter control, sensory function, intellectual ability, and skin integrity are preserved. Patients become paralyzed and often require ventilation and surgery to provide a new opening in the stomach (gastrostomy). Loss of respiratory function is ultimately the cause of death. Incidence and Prevalence Approximately 30,000 patients in the United States currently have ALS. The disease has no racial, socioeconomic, or ethnic boundaries. The life expectancy of ALS patients is usually 3 to 5 years after diagnosis. ALS is most commonly diagnosed in middle age and affects men more often than women. Risk Factors. Risk factors include an inherited genetic defect, which accounts for 5–10% of cases of familial ALS (FALS) in the United States. FALS is linked to a genetic defect on chromosome 21. This gene codes for an enzyme called superoxide dismutase (SOD), an antioxidant that protects motor neurons from free radical damage (i.e., molecules introduced to the body, or produced by body processes that interact and cause cellular damage). More than 60 different mutations that cause SOD to lose its antioxidant properties have been found. However, only 40% of familial ALS cases are linked to SOD mutations, so there may be other unknown genetic defects involved. In the United States, 90–95% of ALS cases are sporadic. Sporadic ALS appears to be increasing worldwide. The causes are not clear, yet some evidence suggests that the immune system may be involved. Excessive levels of glutamate can overstimulate motor neurons and cause them to die. Glutamate is one of the most important neurotransmitters for healthy brain function. Neurotransmitters are chemicals that transmit signals from one nerve to another. In Guamanian ALS, a dietary neurotoxin is the risk factor. The suspected neurotoxin is an amino acid (BMAA) found in the seed of the cycad Cyas cirinalis, a tropical plant found in Guam, which was used to make flour and was a major dietary component during the 1950s and the early 1960s, when this type of ALS had an exceptionally high incidence. Causes. The cause of ALS is not completely understood. Researchers and physicians suspect viruses, neurotoxins (especially in Guamanian ALS), heavy metals, DNA defects (especially in familial ALS), immune system abnormalities, and enzyme abnormalities. Signs and Symptoms Once a motor neuron degenerates completely, the muscle that it controls no longer receives impulses from the brain. Approximately 60% of ALS patients experience muscle weakness and stiffness as the initial symptom. Usually the first muscles affected are those in the arms and legs. Walking or climbing stairs may be difficult. The patient may drop things, fall, experience muscle cramps, and laugh or cry uncontrollably. The arms and legs may feel especially tired. If the hands are affected, the patient may have difficulty picking up small objects or turning keys. Speech problems, such as slurring, hoarseness, or decreased volume may also occur. Signs and symptoms include the following: Absence of spinal reflexes (areflexia) Loss of muscle tone (hypotonia) Muscle twitching (fasciculations) Muscle wasting (atrophy) The signs that the upper motor neurons are affected include the following: Excessive salivation. Extension of the big toe and abduction of the rest of the toes in response to lightly stroking the sole of the foot (Babinski s sign) Reduced muscle tone, or hypotonia, and rigidity (spasticity) Hyperactive tendon reflexes. Impaired speech (dysarthria) Impaired swallowing (dysphagia) Rapidly alternating muscle contractions and relaxations (clonus) As symptoms progressively worsen, the patient s muscles atrophy causing spasticity, stiffness, abnormal movements, and alterations in gait and manual dexterity. Twitching may occur in the tongue and in affected limbs. The patient may experience muscle pain and muscle cramps. Some patients experience more difficulty swallowing saliva and liquids than solid food. Excessive salivation and difficulty swallowing may cause drooling. When respiratory muscles weaken, the patient may require a ventilator. ALS patients often experience fear, anxiety, and depression. Overview Diagnosis ALS is difficult to diagnose because the symptoms are similar to those of other neuromuscular disorders, many of which are treatable. The diagnosis is usually based on a complete neurological examination and clinical tests. The neurological exam usually shows evidence of muscle weakness (localized or widespread, depending on the extent of the disease). The exam also reveals muscle atrophy. The muscles may be so stiff that when the neurologist moves them, they continue to move abnormally afterward. When the neurologist tests the knee jerk reaction, the movement is abnormally quick. Because ALS affects the skeletal, voluntary muscles, the neurological exam does not reveal abnormalities in the sensory reflexes (i.e., vision, hearing, taste, smell, touch, or bowel and bladder control). Tests Nerve conduction velocity (NCV) and electromyography (EMG) help diagnose nerve and muscle disorders. These tests, which can be used in combination, are often referred to as EMG/NCV studies. NCV is administered before EMG and measures the speed at which nerves transmit electrical signals. During NCV, electrodes are placed on the skin over a nerve that supplies a specific muscle or muscle group. A mild, brief electrical stimulus is delivered through the electrode and the response of the muscle is detected, amplified, and displayed. The strength of the signal is also measured. Neurological conditions can cause the NCV to slow down or to be slower on one side of the body. The strength of the response also provides clues to help with diagnosis and to determine the extent of the disease. EMG measures nerve impulses within the muscles. Tiny electrodes are placed in the muscles in the arms and legs and the electronic responses are observed using an instrument that displays movement of an electric current (oscilloscope). As muscles contract, they emit a weak electrical signal that can be detected, amplified, and tracked, providing information about how well the muscles are working. These responses are abnormal in cases of ALS. Tests may be performed to rule out other neurological disorders. Magnetic resonance imaging (MRI scan) may be used to rule out spinal cord disease. Blood tests may be done to detect the presence of heavy metals such as lead in the blood. Laboratory tests may detect abnormal proteins or hormone levels associated with other neurological diseases. A lumbar puncture or spinal tap may be performed to analyze the cerebrospinal fluid for genetic abnormalities (e.g., viral, autoimmune, neurotoxic). Overview Treatment There is no cure for ALS. Treatment focuses on relieving symptoms and maintaining an optimal quality of life. Treatment is based on individual therapy and the continual adaptation of medications. Riluzole (Rilutek®) is one of the few drugs effective against ALS and may prevent progression and prolong life for a few months or so. Riluzole decreases the release of glutamate. Side effects include the following: Dizziness. Elevated liver enzymes. Reduced leukocytes in the blood (granulocytopenia) Weakness (asthenia) Baclofen (Lioresol®) or tizanadine (Zanaflex®) may relieve spasticity. Side effects include increased weakness, sedation, and dizziness. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen or naproxen may relieve general discomfort. Due to potentially severe gastrointestinal and cardiovascular side effects, NSAIDs should only be used as instructed. Tramadol (Ultram®) is often prescribed for pain relief. Physical therapy is an important part of treatment and helps to relieve cramping and muscular pain. Passive stretching helps to avoid permanent contraction of muscles (contractures) that may cause joint problems. Other therapies, such as occupational therapy and speech therapy, are also used in treatment. ALS patients require a diet of high-energy foods that are easy to swallow. Patients may benefit from a nutritionist. If the patient is not able to maintain adequate nutrition, a percutaneous endoscopic gastrostomy (PEG), or feeding tube, is usually inserted. Some ALS patients may also need pulmonary consultants and respiratory therapists to assist breathing. Fewer than 5% of patients use long-term ventilation support. Depression is very common among ALS patients. Antidepressant medication and counseling can help patients and their families cope. Prognosis. ALS is a terminal illness. Fifty percent of patients die within 3 years of diagnosis; 20% live 5 years; and 10% live 10 years. Hospice care can provide comfort and dignity to patients and their loved ones. Resources for patients and caregivers are available from the ALS Association and the ALS Division of t.")
10
Diagnostics EMG / NCV studies
- NCV is administered before EMG and measures the speed at which nerves transmit electrical signals - EMG measures nerve impulses within the muscles Muscle Biopsy Tests to rule out other neurological disorders - MRI may be used to rule out spinal cord diseases - Blood tests may be done to detect the presence of heavy metals such as lead, abnormal proteins or hormone levels associated with other neurological diseases - Lumbar puncture to analyze the cerebrospinal fluid for genetic abnormalities (e.g., viral, autoimmune, neurotoxic)
")
11
Renowned scientist Stephen Hawking suffers from amyotrophic lateral sclerosis

12
** Ability to think or reason remain intact !
Late Stage… As disease progresses and more muscle groups are affected, the person becomes progressively incapacitated When respiratory muscles weaken, the client will require a ventilator Percutaneous Endoscopic Gastrostomy (PEG) or feeding tube ALS patients often experience fear, anxiety, & depression ** Ability to think or reason remain intact !
 or feeding tube. ALS patients often experience fear, anxiety, & depression. ** Ability to think or reason remain intact !")
13
Collaborative Goals Focus on maintaining quality of life
Control symptoms Prevent complications Provide adaptive devices to increase mobility and self-care

14
Collaborative Team Physical therapy helps to relieve cramping and muscular pain. Passive stretching helps to avoid permanent contraction of muscles (contractures) that may cause joint problems Dietician ensures diet of high-energy foods that are easy to swallow Splints, braces, and wheelchairs are used to help with mobility Occupational and Speech therapy as their motor control gradually deteriorates
 that may cause joint problems. Dietician ensures diet of high-energy foods that are easy to swallow. Splints, braces, and wheelchairs are used to help with mobility. Occupational and Speech therapy as their motor control gradually deteriorates.")
15
Medication Riluzole (Rilutek®) is one of the few drugs effective against ALS and may prevent progression and prolong life for a few months or so…
 is one of the few drugs effective against ALS and may prevent progression and prolong life for a few months or so…")
16
Medication - Baclofen (Lioresal) - Damtrolene sodium (Dantrium)
Antispasmodics: - Baclofen (Lioresal) - Damtrolene sodium (Dantrium) - Diazepam (Valium)
 - Damtrolene sodium (Dantrium) - Diazepam (Valium)")
17
Meds… Nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen or naproxen may relieve general discomfort Tramadol (Ultram®) is often prescribed for pain relief
 is often prescribed for pain relief.")
18
Depression Very common
Antidepressant medication and counseling can help patients and their families cope

19
Affairs in Order… While it may be emotionally difficult, it is important for clients and caregivers to make informed, carefully considered decisions regarding the future while the patient is capable of making his or her contribution to a planned course of action Patients and their family members should discuss and consider issues such as legal concerns, home care, assisted care, and institutionalization Draw up wills and other important documents as early as possible to avoid legal problems later on, when the patient may be unable to represent his or her own interests Legal assistance may be necessary if the patient encounters discrimination over insurance or employment.

20
Prognosis Fifty percent of patients die within 3 years of diagnosis
20% live 5 years 10% live 10 years Hospice care can provide comfort and dignity to patients and their loved ones

Similar presentations

>")
 (MND) are a group of neurological disorders that selectively affect motor neurons.>")




 LaTasha Wilson Nate Jr.. Pathophysiology of MS In MS, the body’s own defense system attacks myelin, the fatty substance that surrounds.>")
>")




>")

 is an inflammatory disease of the Central Nervous System (CNS) - that's the brain and spinal cord. Predominantly,>")

 Sarah Belair and Hannah McLaughlin.>")

