Download presentation
Presentation is loading. Please wait.

1
Medical Device Regulation - the next twenty years ?-
Mike Kreuzer OBE Executive Director, Technical and Regulatory

2
The medical technology sector The current regulatory system
The Commission proposal But first ABHI - what we do for our members

3
ABHI STRATEGY UK MARKET REGULATION & STANDARDS ETHICS & PRINCIPLES
Advocating policies that allow members to operate in a favorable business environment UK MARKET Policies that support the rapid evaluation, reimbursement and adoption of medical technologies by UK healthcare systems INTERNATIONAL MARKETS Policies to provide an effective gateway to foreign markets REGULATION & STANDARDS Policies for simple and smart regulation, providing patients with safe, effective, high quality and innovative medical technologies ETHICS & PRINCIPLES Policies to ensure business is conducted in the right manner

4
ABHI’s role in Regulation
ABHI founded 1989 to address EU Medical Device Legislation European dimension to ABHI activity in this area Today we: Monitor developments in the regulatory system Influence the regulators in the UK and at European level (with Eucomed) Provide a limited advisory role to members
 Provide a limited advisory role to members.")
5
The Virtuous Circle European Commission MHRA/ Notified Bodies UK Rep
UK Industry (ABHI) Eucomed
 Eucomed.")
6
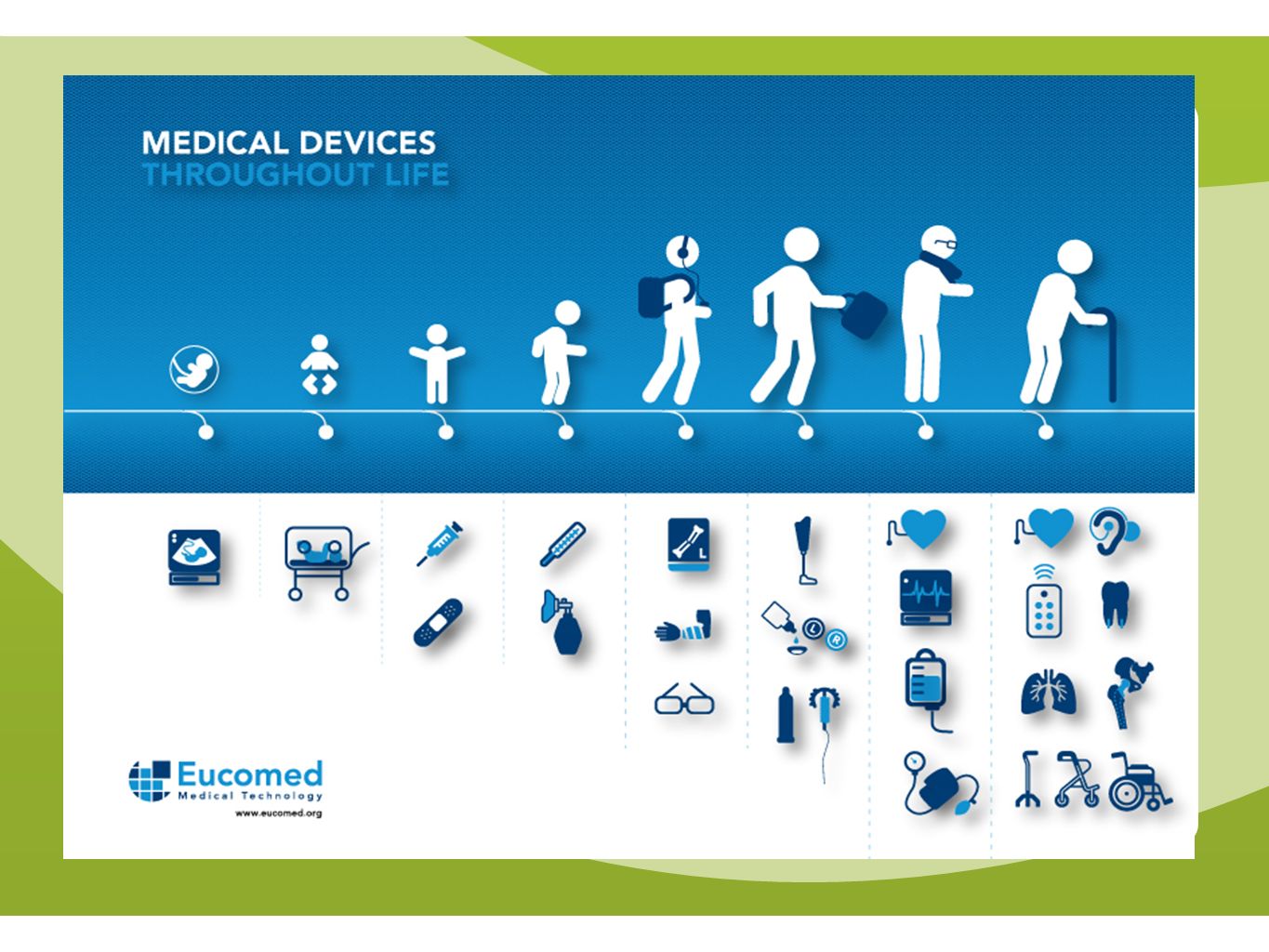
I. The Medical Technology Sector 'It's bigger than you may think'
Please move this slide to after current slide 6 The topic number will change to II

7
The Medical Technology Sector

9
Medical technology industry in the EU
About 22,500 medical technology companies in EU 80% SMEs 95 billion EUR annual sales; 8% re-invested in EU Nearly 500,000 employees > 500,000 products (10,000 generic groups) One new European patent every 38 minutes*
 One new European patent every 38 minutes*")
10
II. The Current Regulatory System
The topic number will change to III

11
Merits of the current system
Stakeholders agree Europe has best regulatory system in the world! High level of patient safety Availability of latest technology solutions Appropriate costs and timings Strong innovation capability Countries with advanced healthcare systems like Australia, Canada use EU model US FDA system is increasingly questioned by domestic media and policy makers This was endorsed by a Commission Communication in mid 2011 Keep merits & basic structure, eliminate the weaknesses Change strong countries to Countries with well developed HC Systems

12
Weaknesses of the current system
Fragmentation: divergent interpretations and applications of rules across EEA Regulatory gaps for certain products: Scope, reprocessing Lack of transparency Shortcomings in implementation Market surveillance / post-market controls Vigilance Functioning of Notified Bodies Damaged confidence in safety the system: PIP and MoM Add slide after this one - text is as follows Immediate Measures called for following PIP Intended to bring forward key measures before final implementation Mainly concern NBs including Regulation due end 2012 Also Recommendation on unannounced inspections

13
The consultation 2008-2011 2008 - initial Recast Paper
2009 – changes in the Commission 2010 – the Exploratory Process 2011 – Commission Communication PIP and HIP the MDR Proposal Add slide after this one - text is as follows Immediate Measures called for following PIP Intended to bring forward key measures before final implementation Mainly concern NBs including Regulation due end 2012 Also Recommendation on unannounced inspections

14
Immediate Measures following PIP
Intended to bring forward key measures before implementation date Mainly concerns NBs including a Regulation due end 2012 Also, Recommendation on unannounced inspections And there will be more Add slide after this one - text is as follows Immediate Measures called for following PIP Intended to bring forward key measures before final implementation Mainly concern NBs including Regulation due end 2012 Also Recommendation on unannounced inspections

15
What we want... Access for patients to life-saving technologies
Safety for patients: PIP never again Access for patients to life-saving technologies Move this slide to come after current slide 15

16
III. The Commission Proposal
Add slide Co-decision process started 26 September Parliament / Council (Member States) and Commission have to agree BUT much could change during this process We now have limited influence BUT we must stay close to - MEPs - National Governments AND those who might otherwise influence them e.g. HCPs
 and Commission have to agree. BUT much could change during this process. We now have limited influence BUT we must stay close to. - MEPs. - National Governments. AND those who might otherwise influence them e.g. HCPs.")
17
The Commission Proposal
We ask three questions of each proposed measure: Does it increase Patient Safety (avoid PIP)? Does it maintain or improve the current access patients and doctors have to life-saving technologies? Does it encourage innovation (sustainable healthcare systems) ?
 Does it maintain or improve the current access patients and doctors have to life-saving technologies Does it encourage innovation (sustainable healthcare systems)")
18
Key elements of the proposal: do they deliver?
Notified Bodies (national coherence, monitoring) P Controls (e.g. unannounced visits) Vigilance (coordination, coherent action) Transparency (databank; public info) Traceability / UDI Clinical Data Governance (coordination by DG SANCO) Scrutiny procedure Q Stakeholder involvement - Reprocessing New legal instrument & scope
 P. Controls (e.g. unannounced visits) Vigilance (coordination, coherent action) Transparency (databank; public info) Traceability / UDI. Clinical Data. Governance (coordination by DG SANCO) Scrutiny procedure. Q. Stakeholder involvement. - Reprocessing. New legal instrument & scope.")
19
Improvements in Notified Bodies (NBs)
Control and oversight largely on voluntary and national approaches lack of transparency, trust and legal certainty Current system More rigorous designation, audit and control by Member States and Commission Member States fees for designation and monitoring of NBs NB enhanced compliance powers – right and duty to carry out: periodic NB audits, unannounced inspections, physical or laboratory testing on MDs, certificate suspensions, withdrawals or restrictions EC proposal P

20
P Vigilance Current system EC proposal
Lack of coordinated exchange of information on reported incidents Considerable variations re responses to incidents Duplication of efforts & increased inequalities re health protection Current system Better coordination between national surveillance authorities Centralized reporting Empowerment of healthcare professionals and patients to report serious incidents at Member State level EC proposal Creation of EU database for centralisation of notifications and coordination of the EC P

21
Transparency and Traceability/UDI
Current system Confidentially requirements seen as too restrictive, lack of transparency Decreased level of public trust in the system and CE-marking Extended database on MDs providing more information available on the quality and safety of devices on the market Introduction of UDI system to enhance post-market safety, reduce medical errors, fight against counterfeiting, enhance purchasing and stock management by hospitals Implant cards EC proposal P

22
UDI – A (rapidly) Emerging Issue
UDI is cross-discipline – Patient Safety / Supply Chain New legislation proposed in 2012 -FDA and EU (in MDD Revision) ‘All devices’ to carry a machine-readable identifier Main purpose: patient safety (traceability) But will be used for ‘commercial’ purposes ABHI can influence development through Eucomed & GHTF Programme will accelerate after PIP BUT key concerns are: -Proliferation of systems -‘reciprocity’ – will healthcare authorities and providers be equipped to interact with industry? 14
 ‘All devices’ to carry a machine-readable identifier. Main purpose: patient safety (traceability) But will be used for ‘commercial’ purposes. ABHI can influence development through Eucomed & GHTF. Programme will accelerate after PIP. BUT key concerns are: -Proliferation of systems. -‘reciprocity’ – will healthcare authorities and providers be equipped to interact with industry 14.")
23
Reinforced clinical evidence
Current system Already legal requirements under current EU law; last improvement in 2007 (Directive 2007/42/EC) Clearer requirements for clinical evidence General rule that class III and implantables should be evaluated on the basis of clinical investigation data New system of centralization of notifications and reporting system for severe adverse event Increased protection of subjects undergoing clinical investigations Extended post-marketing clinical follow-up EC proposal P 21
 Clearer requirements for clinical evidence. General rule that class III and implantables should be evaluated on the basis of clinical investigation data. New system of centralization of notifications and reporting system for severe adverse event. Increased protection of subjects undergoing clinical investigations. Extended post-marketing clinical follow-up. EC proposal. P. 21.")
24
P Governance Stress that this is the HOW rather than the WHAT
Current system Good but suffers from fragmented and implementation EC proposal Improved cooperation and coordination between Member States New Medical Device Coordination Group of MSs EC coordinating role to assist MSs manage the system Increased resources at EU level (DG SANCO, JRC) Stress that this is the HOW rather than the WHAT P
 Stress that this is the HOW rather than the WHAT. P.")
25
New legal instrument & scope
Current system 3 Directives, fragmented implementation Issues related e.g. borderline products or devices for aesthetic purposes 2 Regulations, delegated and implementing acts Wider and clearer scope, e.g. to include implants for aesthetic purposes, devices containing or being made of non-viable human tissues Relabeling and repackaging by parallel importers Distance sales: diagnostics/therapeutics and associated services Clarification re medical software EC proposal P

26
Reprocessing of Single Use Devices
Current system It is not explicitly covered by the current legislation; some labeling requirements EC proposal SCENIHR recommendation followed: reprocessors assigned the same duties as manufacturers; some products will be allowed reprocessing only after appropriate evaluation from EC and MSs; MSs left free to prohibit reprocessing on their territories P

27
Standards & Guidelines
Current system Inefficiencies in development and severe disparities in implementation of guidelines EC proposal Better management of development and harmonized implementation of EU guidance now formal responsibility of the new Medical Device Coordination Group Possibility of ‘Common Technical Specifications’ where no standards exist Caveat on CTSs BUT OK with full stakeholder involvement hence box on Formal Advisory Committee Need for full stakeholder involvement via a formal advisory committee P

28
P - Economic Operators Current system EC proposal
Not all economic operators included Not aligned to New Legislative Framework EC proposal Clearer roles and responsibilities for manufacturers, authorized representatives, importers and distributors Inclusion of diagnostic services and internet sales ‘Qualified Person’ concept introduced to strengthen product safety Problems may arise when considered across all organisational models and supply chain structures P -

29
P - Fees - Current system EC proposal
Industry pays government differently in each Member State in a variety of ways EC proposal Now explicitly expressed – national approaches Appropriate and sustainable funding model that demonstrates benefits for both the regulator and the regulated - P -

30
Stakeholder Involvement
Current system Medical Devices Experts Group (MDEG) open to representatives from valid stakeholders (industry, patients, physician groups) EC proposal No explicit reference to a stakeholder advisory committee MDEG should be kept and given explicit reference in the legislation Q -
 open to representatives from valid stakeholders (industry, patients, physician groups) EC proposal. No explicit reference to a stakeholder advisory committee. MDEG should be kept and given explicit reference in the legislation. Q. -")
31
? - Classification - Current system EC proposal
Risk-based classification: Class I (lower), Class IIa, Class IIb, Class III (Higher) Merger of AIMD and MDD texts; devices covered by AIMD become de facto Class III New rules (Class III): certain devices incorporating nanomaterial, devices for aphaeresis, devices ingested, inhaled or administered rectally or vaginally …. EC proposal Already safe products should not be unnecessarily burdened with increased bureaucracy and costs - ? -
, Class IIa, Class IIb, Class III (Higher) Merger of AIMD and MDD texts; devices covered by AIMD become de facto Class III. New rules (Class III): certain devices incorporating nanomaterial, devices for aphaeresis, devices ingested, inhaled or administered rectally or vaginally …. EC proposal. Already safe products should not be unnecessarily burdened with increased bureaucracy and costs. - -")
32
Early Scientific Advice
Current system Lacks early independent scientific advice on medical technology to Member States, European Commission and innovators EC proposal Mention of Joint Research Centre and Member State Experts But no ability to offer early scientific advice No availability of 'official' advice during development process This was in February draft BUT was excluded in final version due to legal concerns Greater access at EU level to sound independent scientific advice would greatly benefit MedTech SMEs - ? 30

33
Major Concern: Scrutiny of certain conformity assessments
Current system Not included in the current legislation Medical Device Coordination Group (MDCG) to oversee in exceptional cases the work of NBs for new class III devices in case of novel technologies, specific public health threats or uneven evaluation by a NB NB notified Commission of all class III conformity assessment applications MDCG’s comments made public in summary EC proposal Add-on to existing approval process = bureaucratic burden without safety gain Delays between 6 months up to 1, 2, 3,… (?) years - Q 31
 to oversee in exceptional cases the work of NBs for new class III devices in case of novel technologies, specific public health threats or uneven evaluation by a NB. NB notified Commission of all class III conformity assessment applications. MDCG’s comments made public in summary. EC proposal. Add-on to existing approval process = bureaucratic burden without safety gain. Delays between 6 months up to 1, 2, 3,… ( ) years. - Q. 31.")
34
32

35
What’s been strengthened
More rigorous designation and audit of Notified Bodies More vigilance and coordination between national surveillance authorities More traceability, UDI, and implant cards Clearer requirements for clinical evidence More effective governance structure of Member States Wider and clearer scope Increased regulation of reprocessing of single-use devices More harmonized guidance and fully stakeholders’ involvement Clearer roles and responsibilities for economic operators

36
Medical Device Regulatory System
How it will affect you? A tougher regime but based on the same principles Higher risk products: more evidence, more scrutiny? Fees payable to MHRA? Higher Notified Body fees Cost of UDI Enhanced control of importers and distributors

37
Six steps to a smarter legal framework for medical devices
1 2 3 Only the best Notified Bodies One approach to vigilance and market surveillance Strengthened harmonized standards Consistent implementation of guidelines Increased transparency An integrated approach: Better coordination and management 4 5 6 35

38
THANK YOU

39
Merits of EU system - Examples
Move these slides to the Annex and relate to FDA slide already there 13

40
Merits of EU system - Examples

41
Parallels between EU MDD & US FDA systems
High level of public health protection P Risk-based approaches Regulatory approach Decentralized under 27 MS National-level agencies Centralized under 1 Country-level Agency Overall responsibility for products on the market National Competent Authorities (CA) FDA Class III reserved for highest risk Responsibility for Pre-market approval for highest risk Class III NB under supervision of CA Lighter controls for lowest risk Active Post Marketing surveillance Periodic auditing of QMS Recognition of international standards to demonstrate safety, performance & quality
 FDA. Class III reserved for highest risk. Responsibility for Pre-market approval for highest risk Class III. NB under supervision of CA. Lighter controls for lowest risk. Active Post Marketing surveillance. Periodic auditing of QMS. Recognition of international standards to demonstrate safety, performance & quality.")
43
ANNEX

44
The current regulatory system
Implemented at national level Notified Bodies (NB), designated and overseen by national ‘Competent Authorities’ Pre-market approval system for high-risk devices Controls of manufacturers and products before launch & throughout lifetime of product
, designated and overseen by national ‘Competent Authorities’ Pre-market approval system for high-risk devices. Controls of manufacturers and products before launch & throughout lifetime of product.")
45
The current regulatory system
Key elements: Essential Requirements (ER) to demonstrate safety & performance = to be met by all devices; ER globally accepted as THE high level requirements of safety for devices High clinical evidence across all classes of devices, not just for high risk devices Conformity Assessment Procedure = clinical studies & data to proof safety and performance of products and quality system of the manufacturing process; all audited by NB Additional ‘Design dossier examination’ for high risk devices High level of inspection of the manufacturer (by Notified Body & Authorities) Market Surveillance (preventive) & Vigilance systems (in case of incidents) Balance between pre and post market activities
 to demonstrate safety & performance = to be met by all devices; ER globally accepted as THE high level requirements of safety for devices. High clinical evidence across all classes of devices, not just for high risk devices. Conformity Assessment Procedure = clinical studies & data to proof safety and performance of products and quality system of the manufacturing process; all audited by NB. Additional ‘Design dossier examination’ for high risk devices. High level of inspection of the manufacturer (by Notified Body & Authorities) Market Surveillance (preventive) & Vigilance systems (in case of incidents) Balance between pre and post market activities.")
46
What does not help…

Similar presentations














 No. 765/2008 on accreditation and market surveillance>")





