Download presentation
Presentation is loading. Please wait.

1
The ACPS’s Process Analytical Technology Subcommittee
Ajaz S. Hussain, Ph.D. Deputy Director Office of Pharmaceutical Science CDER, FDA ACPS Meeting November 28, 2001

2
Objectives of PAT Discussion
To delineate the goals and objectives of the ACPS’s Subcommittee on PAT Enumerate expectations of the ACPS reporting and timeline

3
Outline Overview (Ajaz Hussain) ACPS discussion and recommendations
Background Information July 19, 2001 ACPS Discussion November 16, 2001 FDA Science Board Discussion A Vision for PAT in Pharmaceutical Manufacturing Proposed responsibilities and timelines October 25, 2001 FR Notice on PAT Subcommittee ACPS discussion and recommendations

4
July 19, 2001 ACPS Discussion on Optimal Applications of PAT
Initiate public discussion on application of process analytical chemistry tools in pharmaceutical manufacturing Strong ACPS support to move forward Recommendation to form a PAT Subcommittee Related discussion on “Rapid Microbial Testing” No further development to report at this time

5
FDA Science Board Discussion Nov. 16, 2001
Speakers Janet Woodcock CDER, FDA Doug Dean and Frances Bruttin PricewaterhouseCoopers G. K. Raju MIT Norman Winskills and Steve Hammond Pfizer Ajaz Hussain OPS, CDER, FDA

6
Science Board’s Response
Strong unanimous endorsement of the proposal Would like to support this initiative talks, seminars,… Would like to receive updates on progress Questions from ACPS?

7
Current Status US Drug products are of high quality, BUT
Dr. Woodcock’s presentation summary Current Status US Drug products are of high quality, BUT Increasing trend toward manufacturing-related problems Low manufacturing and QA process efficiency--cost implications Innovation, modernization and adoption of new technologies slowed Introduction of new technologies in facilities not for US market High burden on FDA resources

8
Dr. Woodcock’s presentation summary
How Did We Get Here? System evolved beginning years ago--when sectors of industry lacked rigorous SOPs Science/technology base did not evolve as quickly as in other sectors Empirical GMP standards necessitates stringent scrutiny International conference on Harmonization--consensus based standards (1990’s) Industry--regulatory risk averse
 Industry--regulatory risk averse.")
9
Dr. Woodcock’s presentation summary
Challenges for FDA How to encourage innovation while ensuring high quality Successful adoption of new technologies will IMPROVE overall quality How to successfully shift from empirical to science based standards for manufacturing process quality How to decrease reliance on pre-approval review and physical evaluation How to recruit and train a scientific workforce proficient in application of new technologies

10
Questions for the Science Board
Dr. Woodcock’s presentation summary Questions for the Science Board Are you able to support the approach? What resources do you suggest FDA draw on? Are there additional aspects to regulation of pharmaceutic quality that we should focus on?

11
Measurement Shows Potential for Improvement
100% Cost reduction Time Compression 0% 35 days 3days Best Practice: VA Ratio 50%

12
Benefits - Increased Effectiveness of Compliance Infrastructure
0% 100% Level of Compliance Cost 5 2 Compliance Gain Direct Cost Recovery

13
PROCESS D WITH QC TESTS: Cycle Times including BULK ACTIVE
20 DAYS 15 DAYS BLEND 2: PRE-BLEND FILM COATING BOTTLE PACKAGING GRANULATION STEP CHEMICAL WEIGHING BLEND 1: FINAL BLEND COMPRESS PROCESSING 10 DAYS 15 DAYS QC1 QC3 QC2 21-90 DAYS 60 DAYS MIT PHARMACEUTICAL MANUFACTURING INITIATIVE (PHARMI)
")
14
ON-LINE TECHNOLOGY IMPACTS DOMINANT CYCLE TIMES
On-line LIF, NIR, Data Analysis, etc. MIT PHARMACEUTICAL MANUFACTURING INITIATIVE (PHARMI)
")
15
LOOKING BEYOND THE “AVERAGE”
Lots with Exceptions Lots without Exceptions OVERALL CYCLE TIMES 100 200 300 400 500 600 700 800 LOT NUMBER NEED FOR FUNDAMENTAL TECHNOLOGY MIT PHARMACEUTICAL MANUFACTURING INITIATIVE (PHARMI)
")
16
Impact of Exceptions PERFORMANCE MEASURE VALUE
(Detailed Analysis of 2 Products) PERFORMANCE MEASURE VALUE Average Cycle time days Std dev(Cycle time) > 100 days Exceptions increase cycle time by > 50 % Exceptions increase variability by > 100% Capacity Utilization of “System” LOW NEED FOR FUNDAMENTAL TECHNOLOGY MIT PHARMACEUTICAL MANUFACTURING INITIATIVE (PHARMI)
 PERFORMANCE MEASURE. VALUE. Average Cycle time 95 days. Std dev(Cycle time) > 100 days. Exceptions increase cycle time by > 50 % Exceptions increase variability by > 100% Capacity Utilization of System LOW. NEED FOR FUNDAMENTAL TECHNOLOGY. MIT PHARMACEUTICAL MANUFACTURING INITIATIVE (PHARMI)")
17
PAT Applications at DP Sites
RM Testing (warehouse based) Packaging Components Blending (at-line or on-line) Drying Tableting (potency and CU) Encapsulation (potency and CU) Tablet Coating (coating thickness) Packaged product Equipment cleaning (on line monitoring of CIP) Equipment cleaning (surface monitoring) Note - Less than 15% of applications at US sites
 Packaging Components. Blending (at-line or on-line) Drying. Tableting (potency and CU) Encapsulation (potency and CU) Tablet Coating (coating thickness) Packaged product. Equipment cleaning (on line monitoring of CIP) Equipment cleaning (surface monitoring) Note - Less than 15% of applications at US sites.")
18
The “Don’t Use” Scenario
What: Modern PAT methods not used/developed during product development so not used for routine process control Why: Fear of regulatory delays Wasteful of resources to duplicate method development -“current methods work OK” Concern of “raising the bar” unnecessarily: information generated for one process may be expected from all Issues: Loss of benefit of PAT - improved process information and control

19
The “Don’t Tell” Scenario
What: PAT methods not registered but used in parallel with registered (conventional) methods to gain greater process insight and control Why: Concern over delays in regulatory approval Concern that data may be interpreted inappropriately by regulators. More data will lead to more deviations from “norms” - need to be able to determine which are relevant and which are not Issues: Duplication, inefficiency, environment of “mistrust”
 methods to gain greater process insight and control. Why: Concern over delays in regulatory approval. Concern that data may be interpreted inappropriately by regulators. More data will lead to more deviations from norms - need to be able to determine which are relevant and which are not. Issues: Duplication, inefficiency, environment of mistrust")
20
The “Win - Win” Scenario
What: Modern PAT methods used to gain greater understanding of processes during development, are registered and used as in-process control (and release?) methods PAT methods accepted as alternatives to traditional lab based methods - but not required Why: Methodology understood and accepted by regulators and industry alike Issues: This is where we should all want to get to. Making progress, but we are not there yet……….
 methods. PAT methods accepted as alternatives to traditional lab based methods - but not required. Why: Methodology understood and accepted by regulators and industry alike. Issues: This is where we should all want to get to. Making progress, but we are not there yet……….")
21
How can we create a “Win-Win” Environment?
Dealing with the real or perceived regulatory hurdles: Sponsor joint forums to promote discussion and enhance understanding of the issues and opportunities offered by PAT Develop an effective process for the evaluation of new PATs Develop appropriate guidelines for the development, validation and implementation of new PATs lab based extraction/chromatography rules don’t apply participate in “dummy run” submissions Ensure consistent approach to PAT by Review and Inspection

22
Shift the Manufacturing Paradigm

23
Issue: Need for FDA to Facilitate Introduction of PAT
Ajaz Hussain’s presentation summary Issue: Need for FDA to Facilitate Introduction of PAT Industry is hesitant to introduce PAT in US Regulatory uncertainty/risk leads to “Don’t Tell” or “Don’t Use” practice New Technology = New Questions Method suitability, chemometrics and validation Old products + New technology = New Regulatory Concerns Problems not visible under the current system Mindset: Why change? PAT application will add to current regulatory requirements

24
“Win-Win” Opportunities
Ajaz Hussain’s presentation summary “Win-Win” Opportunities Optimal application of modern process analytical technologies can Improve quality and manufacturing efficiency Reduce the likelihood of scrap/recalls Improve the scientific and engineering basis of many current FDA-Industry debates

25
What Should FDA Do to Facilitate Introduction of PAT?
Ajaz Hussain’s presentation summary What Should FDA Do to Facilitate Introduction of PAT? Eliminate regulatory uncertainty Official position - FDA will accept new technology that is based on “good” science Develop standards for PAT Method suitability and validation Multivariate statistical/computer pattern recognition Critical process control points and specifications Changes OOS….

26
What Should FDA do to Facilitate Introduction of PAT?
Ajaz Hussain’s presentation summary What Should FDA do to Facilitate Introduction of PAT? Define a clear science based regulatory process Current system “adequate for intended use” Introduction of PAT not a requirement Define conditions under which PAT may replace current “regulatory release testing” Process for addressing existing “invisible” problems in marketed products Review and inspection practices International harmonization

27
How Should FDA Facilitate PAT?
Ajaz Hussain’s presentation summary How Should FDA Facilitate PAT? Limited institutional knowledge and experience at FDA Seek input and collaboration Advisory Committee for Pharmaceutical Science - Subcommittee on PAT Industry (individual companies?) Academic Pharmaceutical Engineering and Process Analytical Chemistry programs PQRI
 Academic Pharmaceutical Engineering and Process Analytical Chemistry programs. PQRI.")
28
A Perspective on PAT: One piece of the puzzle
“Vision I can see clearly now” Quality & performance by design + Continuous “real time” monitoring of quality Specifications based on mechanistic understanding of how formulation and process factors impact product performance High efficiency and capacity utilization “Real time” review and inspection from Rockville, White Oak, NJDO,...

29
Key Elements of the Emerging Program on PAT (Draft)
A “general principles” guidance on PAT Articulate an FDA position on PAT Definitions and terminology Outline a regulatory process for introducing PAT Pre- and post approval phases Addressing existing but invisible problems “Team” approach for review and inspection Types of experimental evidence and justification “Alternate” and “Primary” control/test “Direct” and “Correlation-based” control/test Appropriate level of redundancy or backup systems On/In/At-line release testing (parametric release)
")
30
Types of Tests/Controls
“Alternate” control/test A PAT tool validated by comparison to a traditional in-process test using development data and/or data from routine production for a period of time. Traditional in-process test discontinued after sufficient data collected to support validation. On-line blend uniformity using NIR validated by comparison to data obtained on blend samples collected using a “thief” “Primary” control/test A PAT tool is developed and validated on its own merits Accuracy, precision, specificity,…….

31
Types of Tests/Controls (Contd.)
Correlation-based controls/tests Use of chemometrics or pattern recognition methods to identify and develop a correlation between a measurement and product attribute E.g., Prediction of tablet hardness, dissolution rate from NIR spectral fingerprints Validation based on predictive performance only Validation based on predictive performance plus mechanistic justification (causal links)
")
33
Parametric Release & Release Tests
What is “parametric release” When "data derived from the manufacturing process sterility assurance validation studies and from in-process controls are judged to provide greater assurance that the lot meets the required low probability of containing a contaminated unit (compared to sterility testing results from finished units drawn from the lot), any sterility test procedure adopted may be minimal, or dispensed with on a routine basis.” (USP) Need to redefine this term
, any sterility test procedure adopted may be minimal, or dispensed with on a routine basis. (USP) Need to redefine this term.")
34
EMEA's Note for Guidance on Parametric Release (effective since 9/01)
Defines Parametric Release as: " a system of release that gives assurance that the product is of the intended quality based on the information collected during the manufacturing process and on the compliance with specific GMP requirements related to parametric release." In addition, this note extends the Parametric Release concept to other dosage forms.

35
Parametric Release: Dissolution?
Provide a greater assurance (compared to the current dissolution test method) Lot will meet established specification Lot will meet established BA/BE Data derived from Process that utilizes in-process controls that can measure and control all critical variables that effect dissolution Appropriately designed manufacturing process validation studies Validation based on predictive performance plus mechanistic justification (causal links)
 Lot will meet established specification. Lot will meet established BA/BE. Data derived from. Process that utilizes in-process controls that can measure and control all critical variables that effect dissolution. Appropriately designed manufacturing process validation studies. Validation based on predictive performance plus mechanistic justification (causal links)")
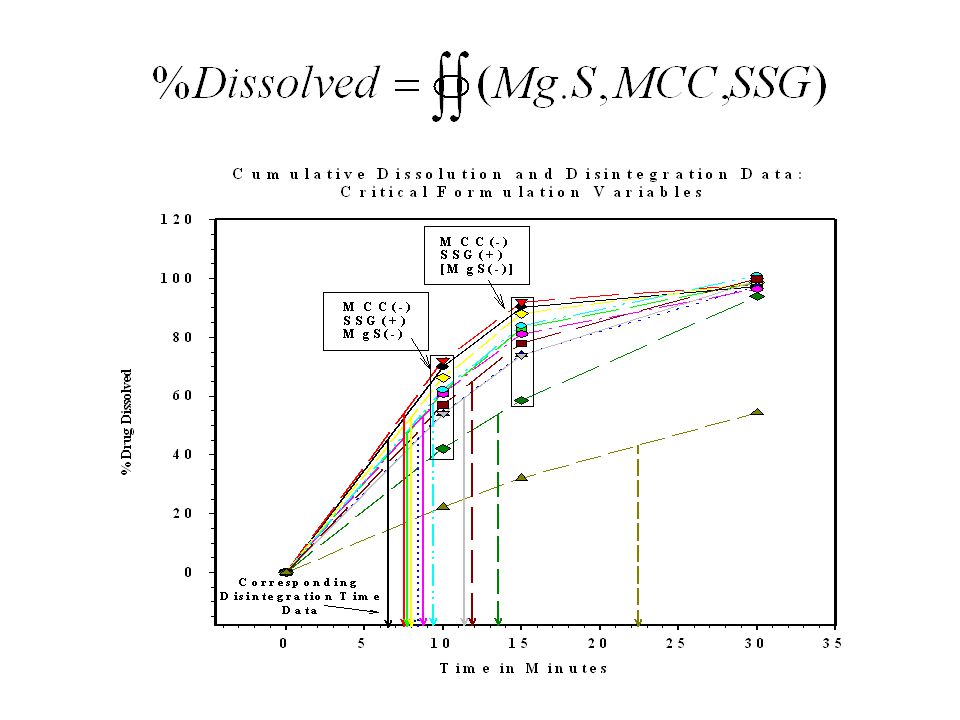
36
Non-homogeneous distribution of magnesium stearate

37
ICH Q6A DECISION TREES #7: SETTING ACCEPTANCE CRITERIA FOR DRUG PRODUCT DISSOLUTION
What specific test conditions and acceptance criteria are appropriate? [IR] YES dissolution significantly affect BA? Develop test conditions and acceptance distinguish batches with unacceptable BA NO Do changes in formulation or manufacturing variables affect dissolution? YES Are these changes controlled by another procedure and acceptance criterion? YES NO NO Adopt appropriate test conditions and acceptance criteria without regard to discriminating power, to pass clinically acceptable batches. Adopt test conditions and acceptance criteria which can distinguish these changes. Generally, single point acceptance criteria are acceptable. aaps Annual Meeting

38
PAT FR Notice Oct. 25, 2001 Request names of qualified individuals
Process analytical chemistry, pharmaceutics, industrial pharmacy, chemical engineering, pharmaceutical analysis, chemometrics, pattern recognition, expert systems, IT, and statistics Report on scientific issues related to application and validation of on-line process technologies (e.g., NIR). Both drug substance and drug product manufacture Feasibility of “parametric” release concept Potential benefits and risks Applications should be received by 11/30/01
. Both drug substance and drug product manufacture. Feasibility of parametric release concept. Potential benefits and risks. Applications should be received by 11/30/01.")
39
Subcommittee should report on (?)
Current status and future trends: PAT in pharmaceutical development and manufacturing Available technologies, capabilities,... Application in US Vs. Non-US plants Perceived and/or real regulatory hurdles General principles for regulatory application Principles of method validation, specifications, OOS Appropriate use and validation of chemometric tools Feasibility of “parametric release” concept (also, redefine) Case study: vibrational spectroscopy (NIR)? Research and training needs (FDA and industry)
 Case study: vibrational spectroscopy (NIR) Research and training needs (FDA and industry)")
Similar presentations






 Overview and Update Robert H. King, Sr. Office of Pharmaceutical.>")



: Status, Challenges and Next Steps Moheb M. Nasr, Ph.D. Office of New Drug Quality Assessment (ONDQA), OPS,>")






 PHARMACEUTICAL MANUFACTURING: NEW TECHNOLOGY OPPORTUNITIES.. G.K.Raju, Ph.D. Executive Director, Pharmaceutical.>")

 and Manufacturing Science Ajaz S. Hussain, Ph.D. Deputy Director, Office of Pharmaceutical Science,>")