Download presentation
Presentation is loading. Please wait.

1
New Drug Update 2013-2014 C. Wayne Weart, Pharm D, FASHP, FAPhA, BCPS
Professor of Clinical Pharmacy and Outcome Sciences South Carolina College of Pharmacy Professor of Family Medicine Medical University of South Carolina Charleston, South Carolina

2
Faculty Disclaimer I am a consultant for Merck in the area of outcomes research.

3
Immunization Update At its October 2012 meeting, the ACIP voted to recommend that healthcare personnel administer a dose of Tdap vaccine to pregnant women during each pregnancy—ideally at between 27 and 36 weeks’ gestation—regardless of the woman’s prior history of receiving Tdap. Reported cases of pertussis have spiked Youngest infants are the most vulnerable Vaccinating the mother during pregnancy can protect the youngest infants. Tdap given at one pregnancy provides insufficient protection for subsequent pregnancies Data support the safety of Tdap for pregnant women and their infants The CDC is expected to publish these recommendations in MMWR

4
FDA Okays Pneumococcal Vaccine for Older Adults
December 20, 2011 The FDA approved the pneumococcal 13-valent conjugate vaccine (manufactured by Wyeth Pharmaceuticals, marketed by Pfizer Inc) for adults aged 50 years and older for the prevention of pneumonia and invasive disease caused by the 13 Streptococcus pneumoniae serotypes contained in the vaccine. The move comes on the heels of the November 16, 2011, meeting of the FDA's Vaccines and Related Biologics Advisory Committee, in which the committee voted 14 to 1 in favor of expanding the indication for Prevnar 13 to adults.
 for adults aged 50 years and older for the prevention of pneumonia and invasive disease caused by the 13 Streptococcus pneumoniae serotypes contained in the vaccine. The move comes on the heels of the November 16, 2011, meeting of the FDA s Vaccines and Related Biologics Advisory Committee, in which the committee voted 14 to 1 in favor of expanding the indication for Prevnar 13 to adults.")
5
CDC Recommends Immunocompromised Adults Get Prevnar 13 Vaccine
June 21, 2012 the Centers for Disease Control and Prevention's Advisory Committee on Vaccine Practices voted 14 to 0 that adults "with AIDS, cancer, organ transplants, advanced kidney disease and other immune-weakening conditions" should be given pneumococcal vaccine Prevnar 13, including those "who've already had Pneumovax 23" The panel has not yet decided if "all adults 50 years old and older should get Prevnar 13."

6
ACIP Recommendations for PCV13 and PPSV23 Use
Adults with specified immunocompromising conditions who are eligible for pneumococcal vaccine should be vaccinated with PCV13 during their next pneumococcal vaccination opportunity. Pneumococcal vaccine-naïve persons. ACIP recommends that adults aged ≥19 years with immunocompromising conditions, functional or anatomic asplenia, CSF leaks, or cochlear implants, and who have not previously received PCV13 or PPSV23, should receive a dose of PCV13 first, followed by a dose of PPSV23 at least 8 weeks later. Subsequent doses of PPSV23 should follow current PPSV23 recommendations for adults at high risk. Specifically, a second PPSV23 dose is recommended 5 years after the first PPSV23 dose for persons aged 19–64 years with functional or anatomic asplenia and for persons with immunocompromising conditions. Additionally, those who received PPSV23 before age 65 years for any indication should receive another dose of the vaccine at age 65 years, or later if at least 5 years have elapsed since their previous PPSV23 dose. MMWR October 12, 2012 / 61(40);
;")
7
ACIP Recommendations for PCV13 and PPSV23 Use
Previous vaccination with PPSV23. Adults aged ≥19 years with immunocompromising conditions, functional or anatomic asplenia, CSF leaks, or cochlear implants, who previously have received ≥1 doses of PPSV23 should be given a PCV13 dose ≥1 year after the last PPSV23 dose was received. For those who require additional doses of PPSV23, the first such dose should be given no sooner than 8 weeks after PCV13 and at least 5 years after the most recent dose of PPSV23. MMWR October 12, 2012 / 61(40);
;")
8
CAPiTA Trial Preliminary Results
The 85,000-patient study in the Netherlands, called CAPiTA, showed that Prevnar 13 prevented invasive pneumococcal disease, meaning infections of Streptococcus pneumoniae bacteria in patients age 65 and older. 45.56% fewer first episodes of vaccine-type CAP among Prevenar 13-vaccinated subjects than in subjects who received placebo (P=0.0006). Secondary objectives, the Prevenar 13 group experienced % fewer first episodes of non-bacteremic/non-invasive vaccine-type CAP (P=0.0067) and % fewer first episodes of vaccine-type IPD (P=0.0005) compared with the placebo group To be reviewed by ACIP and FDA
. Secondary objectives, the Prevenar 13 group experienced % fewer first episodes of non-bacteremic/non-invasive vaccine-type CAP (P=0.0067) and % fewer first episodes of vaccine-type IPD (P=0.0005) compared with the placebo group. To be reviewed by ACIP and FDA.")
9
Influenza Vaccine in Patients with Egg Allergy?
The American College of Allergy, Asthma and Immunology "The very low risk of reacting to the injection is greatly outweighed by the risks associated with the flu." ACAAI recommends that those with a previous history of egg allergy get the injectable vaccine in a medical facility where any allergic emergencies can be recognized and treated if they occur. For those who have had serious reactions after eating eggs, the vaccine should be administered in an allergist's office. In the past, there was concern that because the flu vaccine is grown in eggs, residual protein could trigger a reaction in those with allergies.

10
Influenza Vaccine in Patients with Egg Allergy?
June 21, 2012 The ACIP meeting marked the 1-year anniversary of a change in recommendations that removed egg allergy as a contraindication to influenza vaccination, and it does not appear that the modification affected the rate of allergic reactions, according to data from the Vaccine Adverse Event Reporting System (VAERS).
.")
11
Influenza Vaccine in Patients with Egg Allergy?
June 20, 2013 the ACIP recommended Protein Science's FluBlok for the season in a 13 to 0 vote for patients with a history of egg allergy. A recently approved cell-based flu vaccine Flucelvax, made by Novartis, uses flu viruses grown in mammalian cells rather than chicken eggs and is thought to contain hardly any traces of egg. However, the vaccine seed strain used to make the vaccine is passaged in eggs, meaning it could contain a minuscule amount of albumin.

12
Influenza Vaccines for 2013-14
Influenza formulation changes for the vaccine have been announced. Trivalent vaccine (IIV3) will cover: A/California/7/2009 (H1N1)-like virus; (H3N2) virus antigenically like the cell-propagated prototype virus A/Victoria/361/2011; and B/Massachusetts/2/2012-like virus. Quadrivalent vaccine (IIV4) will also include additional B virus coverage: B/Brisbane/60/2008-like virus Note that the FDA must first approve any changes before they can be made
 will cover: A/California/7/2009 (H1N1)-like virus; (H3N2) virus antigenically like the cell-propagated prototype virus A/Victoria/361/2011; and B/Massachusetts/2/2012-like virus. Quadrivalent vaccine (IIV4) will also include additional B virus coverage: B/Brisbane/60/2008-like virus. Note that the FDA must first approve any changes before they can be made.")
13
Recently-FDA approved Influenza Vaccines for 2013-14 Season
Quadrivalent Live-attenuated Influenza Vaccine (LAIV4)—Flumist Quadrivalent (MedImmune) age 2-49 (2 influenza A and 2 influenza B strains) Quadrivalent Inactivated Influenza Vaccine (IIV4)—Fluarix Quadrivalent (GSK) age 3 and older and Fluzone Quadrivalent (Sanofi) age 6 mo and older both have (2 influenza A and 2 influenza B strains) Cell-culture based inactivated influenza vaccine (ccIIV3)—Flucelvax (Novartis) age 18 and older Recombinant hemagglutinin vaccine (RIV3)—FluBlok (Protein Sciences) age 18-49
—Flumist Quadrivalent (MedImmune) age 2-49 (2 influenza A and 2 influenza B strains) Quadrivalent Inactivated Influenza Vaccine (IIV4)—Fluarix Quadrivalent (GSK) age 3 and older and Fluzone Quadrivalent (Sanofi) age 6 mo and older both have (2 influenza A and 2 influenza B strains) Cell-culture based inactivated influenza vaccine (ccIIV3)—Flucelvax (Novartis) age 18 and older. Recombinant hemagglutinin vaccine (RIV3)—FluBlok (Protein Sciences) age")
14
Existing Influenza Vaccines for 2013-14?
Fluvirin – II3/TIV by Novartis age 4 yrs and up FluLavel – II3/TIV by GSK age 18 yrs and up Afluria – II3/TIV by Merck age 9 yrs and up Fluzone High Dose – II3/TIV by Sanofi age 65 and older Fluzone Interdermal – II3/TIV by Sanofi age 18 to 64 yrs

15
Influenza Vaccine Efficacy
Interim results for the 2013–14 season indicate that vaccination has reduced the risk for influenza-associated medical visits by approximately 60%. (MMWR ) Last year the vaccine was less effective and we now may know why? A British Columbia research team found that mutations in the H3N2 virus occurred when a common strain of the virus chosen for the manufacture of a vaccine was modified. Although this so-called reference strain was initially well-matched to the virus it was intended to protect against, it had to be altered to grow better in chicken eggs to produce the vaccine. It was at this stage in the vaccine-making process that the misstep occurred.
 Last year the vaccine was less effective and we now may know why A British Columbia research team found that mutations in the H3N2 virus occurred when a common strain of the virus chosen for the manufacture of a vaccine was modified. Although this so-called reference strain was initially well-matched to the virus it was intended to protect against, it had to be altered to grow better in chicken eggs to produce the vaccine. It was at this stage in the vaccine-making process that the misstep occurred.")
16
Chemotherapy for Influenza?
When used as a treatment, both neuraminidase inhibitors cut the time to the relief of symptoms -- by about 17 hours for oseltamivir and 14 hours for zanamivir. When used as prophylaxis in people exposed to the flu, both reduced the risk of symptomatic disease but not asymptomatic infection and therefore would have little effect on the risk of transmission during a pandemic. Oseltamivir caused nausea and vomiting and increased the risk of headaches, renal events (decreased creatinine clearance) and psychiatric syndromes (such as confusion and depression). Zanamivir, an inhaled drug, was linked to cases of bronchospasm, but was mostly well tolerated, possibly because it has a low bioavailability. Neither drug was shown to reduce the risk of hospital admission for flu, although there was a slight reduction in self-reported but unverified pneumonia among adults for both drugs. BMJ 2014;348:g2371; g2524; g2545; g2548 from the Cochrane Reviews
 and psychiatric syndromes (such as confusion and depression). Zanamivir, an inhaled drug, was linked to cases of bronchospasm, but was mostly well tolerated, possibly because it has a low bioavailability. Neither drug was shown to reduce the risk of hospital admission for flu, although there was a slight reduction in self-reported but unverified pneumonia among adults for both drugs. BMJ 2014;348:g2371; g2524; g2545; g2548 from the Cochrane Reviews.")
17
FDA approves Flucelvax by Novartis
November 20, 2012 The U.S. Food and Drug Administration announced today the approval of Flucelvax, the first seasonal influenza vaccine licensed in the United States produced using cultured animal cells, instead of fertilized chicken eggs. Flucelvax is approved to prevent seasonal influenza in people ages 18 years and older. The manufacturing process for Flucelvax is similar to the egg-based production method, the virus strains included in the vaccine are grown in animal cells of mammalian origin instead of in eggs. Cell culture technology has already been in use for several decades to produce other U.S. licensed vaccines (polio, rubella and hepatitis A). A “subunit” influenza virus vaccine prepared from virus propagated in Madin Darby Canine Kidney (MDCK) cells.
. A subunit influenza virus vaccine prepared from virus propagated in Madin Darby Canine Kidney (MDCK) cells.")
18
Flublok by Protein Sciences Corporation
Flublok (Influenza Vaccine) Sterile Solution for Intramuscular Injection contains purified HA proteins produced in a continuous insect cell line (expresSF+®) that is derived from Sf9 cells of the fall armyworm, Spodoptera frugiperda, and grown in serum-free medium composed of chemically-defined lipids, vitamins, amino acids, and mineral salts. Flublok is approved for use in persons 18 through 49 years Flublok has a shorter shelf life, with an expiration period of 16 weeks from the production date, as compared to currently available inactivated influenza vaccines which carry an expiration date of June 30 For the influenza season it is formulated to contain 135 mcg HA per 0.5 mL dose, with 45 mcg HA of each of the following 3 influenza virus strains: A/California/7/2009 (H1N1), A/Victoria/361/2011 (H3N2), and B/Wisconsin/1/2010. FDA approval
 Sterile Solution for Intramuscular Injection contains purified HA proteins produced in a continuous insect cell line (expresSF+®) that is derived from Sf9 cells of the fall armyworm, Spodoptera frugiperda, and grown in serum-free medium composed of chemically-defined lipids, vitamins, amino acids, and mineral salts. Flublok is approved for use in persons 18 through 49 years. Flublok has a shorter shelf life, with an expiration period of 16 weeks from the production date, as compared to currently available inactivated influenza vaccines which carry an expiration date of June 30. For the influenza season it is formulated to contain 135 mcg HA per 0.5 mL dose, with 45 mcg HA of each of the following 3 influenza virus strains: A/California/7/2009 (H1N1), A/Victoria/361/2011 (H3N2), and B/Wisconsin/1/2010. FDA approval")
19
ACIP Meeting Fluzone High-Dose was 24.2% more effective in preventing influenza in 32,000 adults aged 65 years or older than regular Fluzone in a large-scale 2 year clinical trial conducted in the US and Canada, vaccine maker Sanofi Pasteur told the Advisory Committee on Immunization Practices of the Centers for Disease Control and Prevention today. The rate of laboratory-confirmed influenza among participants receiving Fluzone High-Dose was 1.43% compared with 1.89% among patients immunized with Fluzone. For the FDA to deem Fluzone High-Dose as superior, the vaccine needed to demonstrate a relative efficacy rate of at least 9.1%. It achieved a rate more than twice that — RRR=24.2%, ARR = 0.46%, NNT 218

20
H5N1 Avian Influenza Vaccine
Nov 22, 2013 Vaccine to supplement National Stockpile, not intended for commercial availability but it is intended to be made available to the public in a pandemic outbreak. The vaccine, Influenza A (H5N1) Virus Monovalent Vaccine, Adjuvanted, is for use in people 18 years of age and older who are at increased risk of exposure to the H5N1 avian influenza virus.
 Virus Monovalent Vaccine, Adjuvanted, is for use in people 18 years of age and older who are at increased risk of exposure to the H5N1 avian influenza virus.")
21
H5N1 Avian Influenza Vaccine
Most avian influenza A viruses do not infect people. However some viruses, such as H5N1, have caused serious illness and death in people outside of the U.S., mostly among people who have been in close contact with infected and ill poultry. When people do become infected with H5N1, about 60 percent die, according to the World Health Organization.

22
H5N1 Avian Influenza Vaccine
The vaccine is made using an egg-based manufacturing process, which is also used for ID Biomedical Corporation’s seasonal influenza vaccine, FluLaval. It contains the adjuvant AS03, an oil-in-water emulsion to enhance the immune response of the vaccinated individual. The adjuvant makes it possible to use a small amount of influenza protein per dose of vaccine to elicit the desired immune response in an individual to prevent influenza disease. Reducing the amount of influenza protein per dose.

23
H5N1 Avian Influenza Vaccine
The H5N1 component and the AS03 adjuvant component are supplied in two separate vials, which must be combined prior to use. The vaccine is administered via intramuscular injection in two doses, 21 days apart. Safety data comes from approximately 3,400 adults 18 years of age and older Muscle aches, headache, fatigue and injection site pain, redness and swelling were common.

24
Meningitis type B Meningitis type B is responsible for about a third of U.S. meningitis cases, but is the only strain not currently preventable by an FDA-approved vaccine. In the last year MenB has infected more than a dozen students at Princeton, UC-Santa Barbara, and Drexel. MenB is a potentially deadly disease which is easily misdiagnosed and can kill within 24 hours of onset. About one in 10 of those who contract the disease will die despite appropriate treatment. Up to one in five survivors may suffer from devastating, life-long disabilities such as brain damage, hearing impairment or limb loss.

25
Meningitis type B Vaccine
Bexsero®, a multi-component Meningococcal B (MenB) vaccine (recombinant, adsorbed) suspension for injection 0.5 ml pre-filled syringe by Novartis is under review at the FDA Safety and efficacy have been shown through clinical trials involving more than 8,000 people including infants, children, adolescents and adults. Available in Canada, EU and Australia NOT YET FDA Approved
 vaccine (recombinant, adsorbed) suspension for injection 0.5 ml pre-filled syringe by Novartis is under review at the FDA. Safety and efficacy have been shown through clinical trials involving more than 8,000 people including infants, children, adolescents and adults. Available in Canada, EU and Australia NOT YET FDA Approved.")
26
Acetaminophen Update January 13, 2011 FDA Drug Safety Communication: Prescription Acetaminophen Products to be Limited to 325 mg Per Dosage Unit - The FDA is asking drug manufacturers to limit the strength of acetaminophen in prescription drug products, which are predominantly combinations of acetaminophen and opioids. This action will limit the amount of acetaminophen in these products to 325 mg per tablet, capsule, or other dosage unit, making these products safer for patients. Drug companies will have three years from the date of publication of the Federal Register Notice (January 14, 2011) to limit the amount of acetaminophen in their oral prescription drug products to 325 mg per dosage unit (see the Federal Register Notice2 Docket number FDA-2011-N ). In addition, a Boxed Warning highlighting the potential for severe liver injury and a Warning highlighting the potential for allergic reactions (e.g., swelling of the face, mouth, and throat, difficulty breathing, itching, or rash) are being added to the label of all prescription drug products that contain acetaminophen.
 to limit the amount of acetaminophen in their oral prescription drug products to 325 mg per dosage unit (see the Federal Register Notice2 Docket number FDA-2011-N ). In addition, a Boxed Warning highlighting the potential for severe liver injury and a Warning highlighting the potential for allergic reactions (e.g., swelling of the face, mouth, and throat, difficulty breathing, itching, or rash) are being added to the label of all prescription drug products that contain acetaminophen.")
27
FDA recommends against prescribing and dispensing prescription combination drug products with more than 325 mg of acetaminophen More than half of manufacturers have voluntarily complied with our request. However, some prescription combination drug products containing more than 325 mg of acetaminophen per dosage unit remain available. In the near future we intend to institute proceedings to withdraw approval of prescription combination drug products containing more than 325 mg of acetaminophen per dosage unit that remain on the market. FDA Jan 14, 2014

28
Top 10 Meds by Prescriptions
Drug 2008 2009 2010 2011 2012 Hydrocodone/aceta 125.5 129.4 132.1 136.7 135.3 Levothyroxine 98.8 100.2 103.2 104.7 107.5 Lisinopril 77.2 83.0 87.6 88,8 90.8 Simvastatin 68.0 84.1 94.4 96.8 86.1 Metoprolol 79.7 76.9 76.6 76.3 78.1 Amlodipine 46.0 52.1 57.8 62.5 66.0 Omeprazole 35.8 45.6 53.5 59.4 65.5 Metformin 51.6 53.8 57.0 59.1 61.6 Albuterol 50.1 54.5 55.1 56.9 61.5 Atorvastatin 58.5 51.7 45.3 43.3 54.9 # of Rx’s (30 and 90 day supply) in millions March 22, 2013 by IMS
 in millions March 22, 2013 by IMS.")
29
DEA Notice of Proposed Rule Making Feb 27, 2014
The Drug Enforcement Administration (DEA) proposes to reschedule hydrocodone combination products from schedule III to schedule II of the Controlled Substances Act. This proposed action is based on a rescheduling recommendation from the Assistant Secretary for Health of the Department of Health and Human Services and an evaluation of all other relevant data by the DEA. If finalized, this action would impose the regulatory controls and administrative, civil, and criminal sanctions applicable to schedule II controlled substances. The DEA will receive comments until April 28, 2014
 proposes to reschedule hydrocodone combination products from schedule III to schedule II of the Controlled Substances Act. This proposed action is based on a rescheduling recommendation from the Assistant Secretary for Health of the Department of Health and Human Services and an evaluation of all other relevant data by the DEA. If finalized, this action would impose the regulatory controls and administrative, civil, and criminal sanctions applicable to schedule II controlled substances. The DEA will receive comments until April 28,")
30
FDA Approves Zohydro ER C-II
10/26/2013 The U.S. Food and Drug Administration today approved Zohydro ER (hydrocodone bitartrate extended-release capsules) by Zogenix for the management of pain severe enough to require daily, around-the-clock, long-term treatment and for which alternative treatment options are inadequate. The first FDA-approved single-entity (not combined with an analgesic such as acetaminophen) and extended-release hydrocodone product.
 by Zogenix for the management of pain severe enough to require daily, around-the-clock, long-term treatment and for which alternative treatment options are inadequate. The first FDA-approved single-entity (not combined with an analgesic such as acetaminophen) and extended-release hydrocodone product.")
31
Hydrocodone bitartrate extended-release capsules – Zohydro ER
ER/LA opioid formulations like Zohydro ER should be reserved for use in patients for whom alternative treatment options are ineffective, not tolerated, or would be otherwise inadequate to provide sufficient management of pain. Zohydro ER is not approved for as-needed pain relief. The approved labeling for Zohydro ER conforms to updated labeling requirements for all ER/LA opioid analgesics announced by the FDA on Sept. 10, 2013.

32
Hydrocodone bitartrate extended-release capsules – Zohydro ER
Extended-release capsules: 10 mg, 15 mg, 20 mg, 30 mg, 40 mg and 50 mg Capsules must be swallowed whole and are not to be chewed, crushed or dissolved. For opioid-naïve and opioid non-tolerant patients, initiate with 10 mg capsules orally every 12 h. To convert to Zohydro ER from another opioid, use available conversion factors to obtain estimated dose. Increase the dose of Zohydro ER in increments of 10 mg every 12 hours every 3 to 7 days as needed to achieve adequate analgesia.

33
EVZIO (Naloxone) Auto-Injector
FDA approved by Kale’o (1-P review) Indication: Naloxone is an opioid antagonist indicated for the emergency treatment of known or suspected opioid overdose, as manifested by respiratory and/or central nervous system depression. (primarily intended for use by the care-giver or a family member) Each dose is 0.4mg of naloxone/0.4 ml Only comes in boxes of two single dose auto-injectors plus a training auto-injector that may be reused
 Indication: Naloxone is an opioid antagonist indicated for the emergency treatment of known or suspected opioid overdose, as manifested by respiratory and/or central nervous system depression. (primarily intended for use by the care-giver or a family member) Each dose is 0.4mg of naloxone/0.4 ml. Only comes in boxes of two single dose auto-injectors plus a training auto-injector that may be reused.")
34
EVZIO (Naloxone) Auto-Injector
A video which demonstrates how to use the auto-injector is available at

35
EVZIO (Naloxone) Auto-Injector
Dosage and Administration: EVZIO is for intramuscular or subcutaneous use only. Administer EVZIO to adult or pediatric patients into the anterolateral aspect of the thigh, through clothing if necessary. Additional doses may be administered every 2 to 3 minutes until emergency medical assistance arrives. In pediatric patients under the age of one, the caregiver should pinch the thigh muscle while administering the dose Seek emergency medical care immediately after use.

36
EVZIO (Naloxone) Auto-Injector
Naloxone is fast onset with peak levels in about 15 min and a half-life of ~1.5 hours (~3.0 hours in neonates) Naloxone will produce rapid reversal of the respiratory and CNS effects of opioids but may only last for 2-3 hours Reversal of partial agonist/antagonists like buprenorphone and pentazocine maybe slow and incomplete especially with buprenorphone and may require larger repeat doses
 Naloxone will produce rapid reversal of the respiratory and CNS effects of opioids but may only last for 2-3 hours. Reversal of partial agonist/antagonists like buprenorphone and pentazocine maybe slow and incomplete especially with buprenorphone and may require larger repeat doses.")
37
EVZIO (Naloxone) Auto-Injector
Patients on long-term opioids will experience opioid withdrawal with nausea, vomiting, sweating, increased heart rate, increased blood pressure, tremors, weakness, body aches and potential seizures as well as increased risk of cardiac events. In neonates, opioid withdrawal may be life-threatening if not recognized and properly treated

38
FDA Drug Safety Communication - Olmesartan
FDA approves label changes to include intestinal problems (sprue-like enteropathy) linked to blood pressure medicine olmesartan medoxomil (Benicar, Azor, Tribenzor) Symptoms of sprue-like enteropathy include severe, chronic diarrhea with substantial weight loss. The enteropathy may develop months to years after starting olmesartan, and sometimes requires hospitalization. Discontinuation of olmesartan has resulted in clinical improvement of sprue-like enteropathy symptoms in all patients. In June 2012, Mayo Clinic researchers published a case series of sprue-like enteropathy associated with olmesartan in 22 patients (Mayo Clin Proc 2012;87:732-8)
 linked to blood pressure medicine olmesartan medoxomil (Benicar, Azor, Tribenzor) Symptoms of sprue-like enteropathy include severe, chronic diarrhea with substantial weight loss. The enteropathy may develop months to years after starting olmesartan, and sometimes requires hospitalization. Discontinuation of olmesartan has resulted in clinical improvement of sprue-like enteropathy symptoms in all patients. In June 2012, Mayo Clinic researchers published a case series of sprue-like enteropathy associated with olmesartan in 22 patients (Mayo Clin Proc 2012;87:732-8)")
39
ACE Inhibitors vs. ARB’s on Outcomes in Patients with Diabetes
Twenty-three trials compared ACEIs with placebo or active drugs (32 827 patients) and 13 compared ARBs with no therapy (controls) (23 867 patients). When compared with controls (placebo/active treatment), ACEIs significantly reduced the risk of all-cause mortality by 13% (RR, 0.87; 95% CI, ), CV deaths by 17% (0.83; ), and major CV events by 14% (0.86; ), including myocardial infarction by 21% (0.79; ) and heart failure by 19% (0.81; ). JAMA Intern Med. doi: /jamainternmed Published online March 31, 2014.
 and 13 compared ARBs with no therapy (controls) ( patients). When compared with controls (placebo/active treatment), ACEIs significantly reduced the risk of all-cause mortality by 13% (RR, 0.87; 95% CI, ), CV deaths by 17% (0.83; ), and major CV events by 14% (0.86; ), including myocardial infarction by 21% (0.79; ) and heart failure by 19% (0.81; ). JAMA Intern Med. doi: /jamainternmed Published online March 31,")
40
ACE Inhibitors vs. ARB’s on Outcomes in Patients with Diabetes
Treatment with ARBs did not significantly affect all-cause mortality (RR, 0.94; 95% CI, ), CV death rate (1.21; ), and major CV events (0.94; ) with the exception of heart failure (0.70; ). Both ACEIs and ARBs were not associated with a decrease in the risk for stroke in patients with DM. Meta-regression analysis showed that the ACEI treatment effect on all-cause mortality and CV death did not vary significantly with the starting baseline blood pressure and proteinuria of the trial participants and the type of ACEI and DM. Conclusion: ACEIs should be considered as first-line therapy to limit excess mortality and morbidity in this population. JAMA Intern Med. doi: /jamainternmed Published online March 31, 2014.
, CV death rate (1.21; ), and major CV events (0.94; ) with the exception of heart failure (0.70; ). Both ACEIs and ARBs were not associated with a decrease in the risk for stroke in patients with DM. Meta-regression analysis showed that the ACEI treatment effect on all-cause mortality and CV death did not vary significantly with the starting baseline blood pressure and proteinuria of the trial participants and the type of ACEI and DM. Conclusion: ACEIs should be considered as first-line therapy to limit excess mortality and morbidity in this population. JAMA Intern Med. doi: /jamainternmed Published online March 31,")
41
FDA Drug Safety Update 8/15/2013
Flouroquinolones may cause disabling peripheral neuropathy symptoms in the arms or legs such as pain, burning, tingling, numbness, weakness, or a change in sensation to light touch, pain or temperature . These symptoms can occur early in treatment and may be permanent. It can occur at any time during treatment with fluoroquinolones and can last for months to years after the drug is stopped or be permanent. Patients using fluoroquinolones who develop any symptoms of peripheral neuropathy should tell their health care professionals right away.

42
Digoxin in Heart Failure : ACC/AHA Heart Failure Guidelines 2013
Class IIa Digoxin can be beneficial in patients with HFrEF, unless contraindicated, to decrease hospitalizations for HF. (Level of Evidence: B) Digoxin Intervention Group Trial (N Engl J Med 1997;336:525-33) Doses of digoxin that achieve a plasma concentration of drug in the range of 0.5 to 0.9 ng/mL are suggested, given the limited evidence currently available Circulation. published online June 5, 2013
 Digoxin Intervention Group Trial (N Engl J Med 1997;336:525-33) Doses of digoxin that achieve a plasma concentration of drug in the range of 0.5 to 0.9 ng/mL are suggested, given the limited evidence currently available. Circulation. published online June 5,")
43
Digoxin and Outcomes? New digoxin use and risks of death and HF hospitalization, controlling for medical history, laboratory results, medications, HF disease severity, and the propensity for digoxin use. We also conducted analyses stratified by sex and concurrent β-blocker use. Among 2891 newly diagnosed patients with systolic HF, 529 (18%) received digoxin. During a median 2.5 years of follow-up, incident digoxin use was associated with higher rates of death (14.2 versus 11.3 per 100 person-years) and HF hospitalization (28.2 versus 24.4 per 100 person-years). In multivariable analysis, incident digoxin use was associated with higher mortality (hazard ratio, 1.72; 95% confidence interval, 1.25–2.36) but no significant difference in the risk of HF hospitalization (hazard ratio, 1.05; 95% confidence interval, 0.82–1.34). Results were similar in analyses stratified by sex and β-blocker use. Circ Cardiovasc Qual Outcomes. 2013;6:
 received digoxin. During a median 2.5 years of follow-up, incident digoxin use was associated with higher rates of death (14.2 versus 11.3 per 100 person-years) and HF hospitalization (28.2 versus 24.4 per 100 person-years). In multivariable analysis, incident digoxin use was associated with higher mortality (hazard ratio, 1.72; 95% confidence interval, 1.25–2.36) but no significant difference in the risk of HF hospitalization (hazard ratio, 1.05; 95% confidence interval, 0.82–1.34). Results were similar in analyses stratified by sex and β-blocker use. Circ Cardiovasc Qual Outcomes. 2013;6:")
44
Digoxin after 230 years? The Editorial by Dr. Opie entitled “Digitalis, Yesterday and Today, But Not Forever” concludes: “This conclusion is the opposite of what the earlier studies favoring digoxin use in the bygone era of imperfect therapy for HF had found, with the new conclusion that therapy for HF that includes β-blockade and full angiotensin-II modulation dispenses with the need for taking the risks of adding digoxin therapy. The data at our disposal, taking into account the current study, allow us to seriously question the advice on digoxin given by both the current and influential guidelines, European and American.” Circ Cardiovasc Qual Outcomes. 2013;6:

45
Aspirin in Patients with Heart Failure
A retrospective cohort study of 1476 patients (mean age 70.4±12.4 years, 63% male) attending a HF disease management program examined aspirin use at baseline and its association with mortality and HF hospitalization (60.4%) were prescribed aspirin (75mg/day in 92.8%). Median follow-up time was 2.6 [0.8:4.5] years. Over the follow-up period, 464 (31.4%) patients died. In adjusted analysis, low-dose aspirin use was associated with reduced mortality risk compared to non-aspirin use (HR=0.58, 95% CI 0.46–0.74). Low-dose aspirin use was associated with reduced risk of HF hospitalization compared to non-aspirin use in the total population (adjusted HR=0.70, 95% CI 0.54–0.90). /CIRCHEARTFAILURE (on-line )
 attending a HF disease management program examined aspirin use at baseline and its association with mortality and HF hospitalization. 892 (60.4%) were prescribed aspirin (75mg/day in 92.8%). Median follow-up time was 2.6 [0.8:4.5] years. Over the follow-up period, 464 (31.4%) patients died. In adjusted analysis, low-dose aspirin use was associated with reduced mortality risk compared to non-aspirin use (HR=0.58, 95% CI 0.46–0.74). Low-dose aspirin use was associated with reduced risk of HF hospitalization compared to non-aspirin use in the total population (adjusted HR=0.70, 95% CI 0.54–0.90) /CIRCHEARTFAILURE (on-line )")
46
Aspirin in Patients with Heart Failure
In adjusted analysis, there was no difference in mortality or HF hospitalization between high-dose aspirin users (>75mg/day) and non-aspirin users. Conclusions— In this study low-dose aspirin therapy was associated with a significant reduction in mortality and morbidity risk over long-term follow-up. /CIRCHEARTFAILURE (on-line )
 and non-aspirin users. Conclusions— In this study low-dose aspirin therapy was associated with a significant reduction in mortality and morbidity risk over long-term follow-up /CIRCHEARTFAILURE (on-line )")
47
OTC Nasacort Allergy 24hr Nasal Spray
Sanofi/Chattem announced that Nasacort (triamcinolone acetonide) Allergy 24hr Nasal Spray is now available over-the-counter (OTC) to relieve a range of seasonal and year-round nasal allergy symptoms, including nasal congestion, in adults and children >2 years of age. Nasacort spray in 60 (~$12) and 120 (~$18) metered dose sprays. (55mcg/spray which is the same as the prescription (vs ~$58.00 for the generic Rx) Nasacort was approved for the switch from prescription to OTC by the FDA on October 11, 2013.
 Allergy 24hr Nasal Spray is now available over-the-counter (OTC) to relieve a range of seasonal and year-round nasal allergy symptoms, including nasal congestion, in adults and children >2 years of age. Nasacort spray in 60 (~$12) and 120 (~$18) metered dose sprays. (55mcg/spray which is the same as the prescription (vs ~$58.00 for the generic Rx) Nasacort was approved for the switch from prescription to OTC by the FDA on October 11,")
48
FDA Approves OTC Nexium 24 HR by Pfizer
March 28, 2014 The FDA approved Nexium 24 HR (esomeprazole magnesium 22.3 mg equivalent to esomeprazole base 20 mg ) for the treatment of frequent heartburn after Pfizer acquired the rights to market the OTC version from Astra Zeneca for $250 million and royalties based upon sales. Nexium 24 HR joins Prilosec OTC, Prevacid 24 HR and Zegerid OTC all QD for 14 days for treatment of frequent heartburn
 for the treatment of frequent heartburn after Pfizer acquired the rights to market the OTC version from Astra Zeneca for $250 million and royalties based upon sales. Nexium 24 HR joins Prilosec OTC, Prevacid 24 HR and Zegerid OTC all QD for 14 days for treatment of frequent heartburn.")
49
NSAID’s and Atrial Fibrillation?
Data from the population-based follow-up study, the Rotterdam Study comprised of 8423 participants without atrial fibrillation at baseline (the mean age of the study population was 68.5 years (SD: 8.7) and 58% were women). During a mean follow-up of 12.9 years, 857 participants developed atrial fibrillation. Current use of NSAIDs was associated with increased risk compared with never-use (HR 1.76, 95% CI 1.07 to 2.88). Also, recent use (within 30 days after discontinuation of NSAIDs) was associated with an increased risk of atrial fibrillation compared with never-use (HR 1.84, 95% CI 1.34 to 2.51) adjusted for age, sex and several potential confounders. BMJOpen 2014; 4: e doi: /
 and 58% were women). During a mean follow-up of 12.9 years, 857 participants developed atrial fibrillation. Current use of NSAIDs was associated with increased risk compared with never-use (HR 1.76, 95% CI 1.07 to 2.88). Also, recent use (within 30 days after discontinuation of NSAIDs) was associated with an increased risk of atrial fibrillation compared with never-use (HR 1.84, 95% CI 1.34 to 2.51) adjusted for age, sex and several potential confounders. BMJOpen 2014; 4: e doi: /")
50
FDA Drug Safety Communication- Testosterone Products
Jan 31,2014 The FDA is investigating the risk of stroke, heart attack, and death in men taking FDA-approved testosterone products. Testosterone products are FDA-approved only for use in men who lack or have low testosterone levels (<300ng/dl) in conjunction with an associated medical condition. Examples include failure of the testicles to produce testosterone, because of reasons such as genetic problems or chemotherapy. Other examples include problems with the hypothalamus and pituitary that control the production of testosterone by the testicles. None of the FDA-approved testosterone products are approved for use in men with low testosterone levels who lack an associated medical condition. FDA-approved testosterone formulations include the topical gel, transdermal patch, buccal system (applied to upper gum or inner cheek), and injection.
 in conjunction with an associated medical condition. Examples include failure of the testicles to produce testosterone, because of reasons such as genetic problems or chemotherapy. Other examples include problems with the hypothalamus and pituitary that control the production of testosterone by the testicles. None of the FDA-approved testosterone products are approved for use in men with low testosterone levels who lack an associated medical condition. FDA-approved testosterone formulations include the topical gel, transdermal patch, buccal system (applied to upper gum or inner cheek), and injection.")
51
FDA Drug Safety Communication- Testosterone Products
An observational study of older men in the U.S. Veteran Affairs health system The population was a very specific group of men who had coronary heart disease based upon coronary angiography and had their testosterone levels measured (<300 ng/dL) before they had a myocardial infarction, rather than after. These men received an appropriate prescription and filled it at least once to see what would happen. The investigators ended up with approximately 8700 men, about 1275 of whom had been given testosterone, and they examined the outcomes. They looked at all-cause mortality, strokes, and heart attacks. They found that giving testosterone at recommended levels to men who were candidates for it actually raised their testosterone levels, as might be expected. JAMA. 2013;310:
 before they had a myocardial infarction, rather than after. These men received an appropriate prescription and filled it at least once to see what would happen. The investigators ended up with approximately 8700 men, about 1275 of whom had been given testosterone, and they examined the outcomes. They looked at all-cause mortality, strokes, and heart attacks. They found that giving testosterone at recommended levels to men who were candidates for it actually raised their testosterone levels, as might be expected. JAMA. 2013;310:")
52
FDA Drug Safety Communication- Testosterone Products
The absolute rate of events (all-cause mortality, MI, and ischemic stroke) were 19.9% in the no testosterone therapy group vs 25.7% in the testosterone therapy group, with an absolute risk difference of 5.8% (95% CI, -1.4% to 13.1%) (NNH = 18) at 3 years after coronary angiography. In Cox proportional hazards models adjusting for the presence of coronary artery disease, testosterone therapy use as a time-varying covariate was associated with increased risk of adverse outcomes (hazard ratio, 1.29; 95% CI, 1.04 to 1.58). JAMA. 2013;310:
 were 19.9% in the no testosterone therapy group vs 25.7% in the testosterone therapy group, with an absolute risk difference of 5.8% (95% CI, -1.4% to 13.1%) (NNH = 18) at 3 years after coronary angiography. In Cox proportional hazards models adjusting for the presence of coronary artery disease, testosterone therapy use as a time-varying covariate was associated with increased risk of adverse outcomes (hazard ratio, 1.29; 95% CI, 1.04 to 1.58). JAMA. 2013;310:")
53
FDA Drug Safety Communication- Testosterone Products
A cohort study of the risk of acute non-fatal myocardial infarction (MI) following an initial topical testosterone (TT) prescription (N = 55,593) in a large US health-care database. Compared the incidence rate of MI in the 90 days following the initial prescription (post-prescription interval) with the rate in the one year prior to the initial prescription (pre-prescription interval) (post/pre). Also compared post/pre rates in a cohort of men prescribed phosphodiesterase type 5 inhibitors (PDE5I; sildenafil or tadalafil, N = 167,279), and compared TT prescription post/pre rates with the PDE5I post/pre rates,adjusting for potential confounders PLOS ONE January 2014 Volume 9 Issue
 following an initial topical testosterone (TT) prescription (N = 55,593) in a large US health-care database. Compared the incidence rate of MI in the 90 days following the initial prescription (post-prescription interval) with the rate in the one year prior to the initial prescription (pre-prescription interval) (post/pre). Also compared post/pre rates in a cohort of men prescribed phosphodiesterase type 5 inhibitors (PDE5I; sildenafil or tadalafil, N = 167,279), and compared TT prescription post/pre rates with the PDE5I post/pre rates,adjusting for potential confounders. PLOS ONE January 2014 Volume 9 Issue")
54
FDA Drug Safety Communication- Testosterone Products
Results: In all subjects, the post/pre-prescription rate ratio (RR) for TT prescription was 1.36 (1.03, 1.81). In men aged 65 years and older, the RR was 2.19 (1.27, 3.77) for TT prescription and 1.15 (0.83, 1.59) for PDE5I, and the ratio of the rate ratios (RRR) for TT prescription relative to PDE5I was 1.90 (1.04, 3.49). The RR for TT prescription increased with age from 0.95 (0.54, 1.67) for men under age 55 years to 3.43 (1.54, 7.56) for those aged >75 years (ptrend = 0.03), while no trend was seen for PDE5I (ptrend = 0.18). In men under age 65 years, excess risk was confined to those with a prior history of heart disease, with RRs of 2.90 (1.49, 5.62) for TT prescription and 1.40 (0.91, 2.14) for PDE5I, and a RRR of 2.07 (1.05, 4.11). PLOS ONE January 2014 Volume 9 Issue
 for TT prescription was 1.36 (1.03, 1.81). In men aged 65 years and older, the RR was 2.19 (1.27, 3.77) for TT prescription and 1.15 (0.83, 1.59) for PDE5I, and the ratio of the rate ratios (RRR) for TT prescription relative to PDE5I was 1.90 (1.04, 3.49). The RR for TT prescription increased with age from 0.95 (0.54, 1.67) for men under age 55 years to 3.43 (1.54, 7.56) for those aged >75 years (ptrend = 0.03), while no trend was seen for PDE5I (ptrend = 0.18). In men under age 65 years, excess risk was confined to those with a prior history of heart disease, with RRs of 2.90 (1.49, 5.62) for TT prescription and 1.40 (0.91, 2.14) for PDE5I, and a RRR of 2.07 (1.05, 4.11). PLOS ONE January 2014 Volume 9 Issue")
55
FDA Drug Safety Communication- Testosterone Products
“At this time, FDA has not concluded that FDA-approved testosterone treatment increases the risk of stroke, heart attack, or death. Patients should not stop taking prescribed testosterone products without first discussing any questions or concerns with their health care professionals. Health care professionals should consider whether the benefits of FDA-approved testosterone treatment is likely to exceed the potential risks of treatment. The prescribing information in the drug labels of FDA-approved testosterone products should be followed.”

56
FDA Approves Testosterone Undecanoate Injection
March 6, 2014 the FDA approved Aveed (testosterone undecanoate) injection for the treatment of adult men with hypogonadism (commonly known as Low-T) that is associated with a deficiency or absence of the male hormone testosterone. Aveed is a new prescription medicine indicated to produce serum testosterone levels in the normal range by administration of a single 3-mL (750 mg) intramuscular injection given once at initiation of therapy, at 4 weeks, and then every 10 weeks thereafter.
 injection for the treatment of adult men with hypogonadism (commonly known as Low-T) that is associated with a deficiency or absence of the male hormone testosterone. Aveed is a new prescription medicine indicated to produce serum testosterone levels in the normal range by administration of a single 3-mL (750 mg) intramuscular injection given once at initiation of therapy, at 4 weeks, and then every 10 weeks thereafter.")
57
Testosterone Undecanoate Injection - Aveed
Approval of Aveed is based on data from an 84-week Phase 3 trial of hypogonadal men in the U.S. Men enrolled in the study had an average age of 54 years and a serum total testosterone level of less than 300 ng/dL. In the Phase 3 study, Aveed increased mean serum testosterone levels, maintaining them for up to 10 weeks at steady state (between weeks 14-24). Aveed is approved with a Risk Evaluation and Mitigation System (REMS) requiring prescriber education and certification as well as restricted product distribution.
. Aveed is approved with a Risk Evaluation and Mitigation System (REMS) requiring prescriber education and certification as well as restricted product distribution.")
58
Testosterone Undecanoate Injection - Aveed
Indicated for replacement therapy in adult males for conditions associated with a deficiency or absence of endogenous testosterone, including primary hypogonadism (congenital or acquired) and hypogonadotropic hypogonadism (congenital or acquired). Aveed has a Boxed Warning for serious pulmonary oil microembolism (POME) reactions and anaphylaxis. It should be used in patients who require therapy and in whom the benefits of the product outweigh the serious risks of POME and severe allergic reaction (anaphylaxis). It must be prescribed and administered by trained healthcare providers in a doctor's office, clinic, or hospital. Patients must remain in the administering physician's office or clinic for at least 30 minutes after injection so that short-term reactions may be observed and treated.
 and hypogonadotropic hypogonadism (congenital or acquired). Aveed has a Boxed Warning for serious pulmonary oil microembolism (POME) reactions and anaphylaxis. It should be used in patients who require therapy and in whom the benefits of the product outweigh the serious risks of POME and severe allergic reaction (anaphylaxis). It must be prescribed and administered by trained healthcare providers in a doctor s office, clinic, or hospital. Patients must remain in the administering physician s office or clinic for at least 30 minutes after injection so that short-term reactions may be observed and treated.")
59
Testosterone Undecanoate Injection - Aveed
Other potential adverse effects include worsened prostate enlargement, liver toxicity, peripheral edema, sleep apnea, and venous thrombosis. Shortly after Endo announced the approval, the consumer group Public Citizen called on the FDA to reverse it, citing recent studies that suggest increased cardiovascular risk with testosterone products.

60
What’s New 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol
A New Perspective on LDL–C and/or Non-HDL–C Treatment Goals The panel makes no recommendations for or against specific LDL–C or non-HDL–C targets for the primary or secondary prevention of ASCVD. The Expert Panel was unable to find randomized clinical trial (RCT) evidence to support continued use of specific LDL–C and/or non-HDL–C treatment targets. Circulation. published online November 12,
 evidence to support continued use of specific LDL–C and/or non-HDL–C treatment targets. Circulation. published online November 12,")
61
What’s New 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol
The appropriate intensity of statin therapy should be used to reduce ASCVD risk in those most likely to benefit. Nonstatin therapies do not provide acceptable ASCVD risk reduction benefits compared to their potential for adverse effects in the routine prevention of ASCVD.

62
High- Moderate- and Low-Intensity Statin Therapy (Used in the RCTs reviewed by the Expert Panel)
High-Intensity Statin Therapy Moderate-Intensity Statin Therapy Low-Intensity Statin Therapy Daily dose lowers LDL–C on average, by approximately ≥50% Daily dose lowers LDL–C on average, by approximately 30% to <50% Daily dose lowers LDL–C on average, by <30% Atorvastatin (40†)–80 mg Rosuvastatin 20 (40) mg Atorvastatin 10 (20) mg Rosuvastatin (5) 10 mg Simvastatin 20–40 mg‡ Pravastatin 40 (80) mg Lovastatin 40 mg Fluvastatin XL 80 mg Fluvastatin 40 mg bid Pitavastatin 2–4 mg Simvastatin 10 mg Pravastatin 10–20 mg Lovastatin 20 mg Fluvastatin 20–40 mg Pitavastatin 1 mg )* Specific statins and doses are noted in bold that were evaluated in RCTs. Statins and doses that are approved by the U.S. FDA but were not tested in the RCTs reviewed are listed in italics.
–80 mg. Rosuvastatin 20 (40) mg. Atorvastatin 10 (20) mg. Rosuvastatin (5) 10 mg. Simvastatin 20–40 mg‡ Pravastatin 40 (80) mg. Lovastatin 40 mg Fluvastatin XL 80 mg Fluvastatin 40 mg bid Pitavastatin 2–4 mg. Simvastatin 10 mg. Pravastatin 10–20 mg. Lovastatin 20 mg. Fluvastatin 20–40 mg. Pitavastatin 1 mg. )* Specific statins and doses are noted in bold that were evaluated in RCTs. Statins and doses that are approved by the U.S. FDA but were not tested in the RCTs reviewed are listed in italics.")
63
What’s New 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol
Global Risk Assessment for Primary Prevention This guideline recommends use of the new Pooled Cohort Equations to estimate 10-year ASCVD risk in both white and black men and women. (Controversial?) By more accurately identifying higher risk individuals for statin therapy, the guideline focuses statin therapy on those most likely to benefit. It also indicates, based on RCT data, those high-risk groups that may not benefit. Before initiating statin therapy, this guideline recommends a discussion by clinician and patients.
 By more accurately identifying higher risk individuals for statin therapy, the guideline focuses statin therapy on those most likely to benefit. It also indicates, based on RCT data, those high-risk groups that may not benefit. Before initiating statin therapy, this guideline recommends a discussion by clinician and patients.")
65
Pooled Cohort Risk Assessment Equations Predicts 10-year risk for a first atherosclerotic cardiovascular disease (ASCVD) event Resultss

66
NNT for Statins for 5 Years:
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. NNT for Statins for 5 Years: 10-year risk of CVD events 5-year NNT for CVD events 5-year NNT for myocardial infarction 5-year NNT for stroke 5-year NNT for mortality 5% 160 278 910 * 7.5% 108 186 606 10% 80 140 456 15% 54 94 304 334 20% 40 70 228 250 Abbreviations: CVD, cardiovascular disease; NNT, number needed to treat (to prevent 1 outcome) * no apparent mortality reduction in lowest-risk patients (BMJ 2013 Oct 22;347:f6123) Courtesty: DynaMed
 * no apparent mortality reduction in lowest-risk patients (BMJ 2013 Oct 22;347:f6123) Courtesty: DynaMed.")
67
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults.
The new guidelines have also drawn considerable criticism especially when it comes to primary prevention and the new recommendation for a 10 year CV risk level of 7.5% with the new risk calculator. Ridker and Cook in a Lancet Editorial commend the new guideline for its emphasis on improving and simplifying use of statins. However, they calculate that the risk prediction algorithm used in the guideline has “systematically overestimated” cardiovascular risks, and could therefore lead to overtreatment. Lancet November pp 1680

68
External Validation How many of the 33 million expected to have risk >7.5% actually have risk that is much lower? The Lancet, Volume 382, Issue 9907, Pages

69
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults.
Treating to LDL cholesterol targets is no longer recommended; rather, clinicians should determine whether a patient falls into one of four mutually exclusive high-risk groups and should initiate statin therapy as follows: 1. Patients with clinical atherosclerotic cardiovascular disease (ASCVD) should receive high-intensity (age, <75) or moderate-intensity (age, ≥75) statin therapy 2. Patients with LDL cholesterol levels ≥190 mg/dL should receive high-intensity statin therapy. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]
 should receive high-intensity (age, <75) or moderate-intensity (age, ≥75) statin therapy. 2. Patients with LDL cholesterol levels ≥190 mg/dL should receive high-intensity statin therapy. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]")
70
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults.
3. Patients with diabetes aged 40–75 with LDL cholesterol levels of 70–189 mg/dL and without clinical ASCVD should receive at least moderate-intensity statin therapy (and possibly high-intensity statin therapy when estimated 10-year ASCVD risk is ≥7.5%) 4. Patients without clinical ASCVD or diabetes but with LDL cholesterol levels of 70–189 mg/dL and estimated 10-year ASCVD risk ≥7.5% should receive moderate- or high-intensity statin therapy J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]
 4. Patients without clinical ASCVD or diabetes but with LDL cholesterol levels of 70–189 mg/dL and estimated 10-year ASCVD risk ≥7.5% should receive moderate- or high-intensity statin therapy. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]")
71
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults.
High-intensity statin therapies are atorvastatin (40–80 mg) or rosuvastatin (Crestor; 20–40 mg). Moderate-intensity statin therapies include atorvastatin (10–20 mg), rosuvastatin (5–10 mg), simvastatin (20–40 mg), pravastatin (40–80 mg), and several others With few exceptions, use of lipid-modifying drugs other than statins is discouraged. Lifestyle modification is recommended for all patients, regardless of cholesterol-lowering drug therapy J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]
 or rosuvastatin (Crestor; 20–40 mg). Moderate-intensity statin therapies include atorvastatin (10–20 mg), rosuvastatin (5–10 mg), simvastatin (20–40 mg), pravastatin (40–80 mg), and several others. With few exceptions, use of lipid-modifying drugs other than statins is discouraged. Lifestyle modification is recommended for all patients, regardless of cholesterol-lowering drug therapy. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]")
72
High Potency Statins vs. Simvastatin vs. Simvastatin plus Ezetimibe?
The UK General Practice Research Database reviewed 9597 patients (57% male, mean age of 65 ±13 years) matched study criteria (had survived 30 days after their first acute myocardial infarct (AMI), had not received prior statin or ezetimibe therapy and were started on a statin within 30 days of AMI were included). Primary outcome was all cause mortality Simvastatin (n=6990 (72.8%)); high-potency statin (n=1883, (19.6%)); and ezetimibe/statin combination (n=724 (7.5%)). During a mean follow-up of 3.2 years, there were 1134 (12%) deaths. the study lacked statistical power to determine any mortality effect with ezetimibe. Heart 2014; DOI: /heartjnl
 matched study criteria (had survived 30 days after their first acute myocardial infarct (AMI), had not received prior statin or ezetimibe therapy and were started on a statin within 30 days of AMI were included). Primary outcome was all cause mortality. Simvastatin (n=6990 (72.8%)); high-potency statin (n=1883, (19.6%)); and ezetimibe/statin combination (n=724 (7.5%)). During a mean follow-up of 3.2 years, there were 1134 (12%) deaths. the study lacked statistical power to determine any mortality effect with ezetimibe. Heart 2014; DOI: /heartjnl")
73
Proportional Hazards Ratio for Risk of Death Heart 2014; DOI: 10
Proportional Hazards Ratio for Risk of Death Heart 2014; DOI: /heartjnl HR 95% CI p Value Cohort (vs simvastatin monotherapy) High-potency statin monotherapy 0.72 0.59 to 0.88 <0.001 Ezetimibe/statin combination 0.96 0.64 to 1.43 0.847 Gender (female vs male) 0.84 0.74 to 0.95 0.009 Age (per year) 1.08 1.08 to 1.09 Smoker (yes vs no) 1.44 1.25 to 1.65 Diabetic (yes vs no) 1.13 to 1.83 Further MI during follow-up 1.45 1.32 to 1.60 Cardiovascular drugs (yes vs no) Aspirin 0.57 0.48 to 0.69 β-Blockers 0.68 0.59 to 0.79 ACE-I 0.62 to 0.84 DHP CCB 0.47 to 0.69
 High-potency statin monotherapy to < Ezetimibe/statin combination to Gender (female vs male) to Age (per year) to Smoker (yes vs no) to Diabetic (yes vs no) 1.13 to Further MI during follow-up to Cardiovascular drugs (yes vs no) Aspirin to β-Blockers to ACE-I to DHP CCB to")
74
AHA/ACC Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults.
This guideline represents a paradigm shift for most clinicians and patients. The rationale for abandoning LDL cholesterol targets is that randomized trials showing benefits of statins generally have examined fixed-dose statin therapy, rather than titrated therapy, to achieve prespecified LDL cholesterol goals. Additionally, some drugs that “improve” the lipid profile (a surrogate endpoint) do not improve clinical outcomes, and statins are thought to exert benefit through pleiotropic effects apart from LDL cholesterol–lowering. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]
 do not improve clinical outcomes, and statins are thought to exert benefit through pleiotropic effects apart from LDL cholesterol–lowering. J Am Coll Cardiol 2013 Nov 12; [e-pub ahead of print]")
75
Statins and Erectile Function?
Data presented at the 2014 ACC Meeting in Wash DC The investigators searched for randomized controlled trials that examined the effect of statin therapy on erectile function. They identified 11 such trials in which men completed the International Inventory of Erectile Function survey, which consists of five questions, each scored on a five-point scale, where low values represent poor sexual function a total of 647 men, an average age of 57.8 years and who had received statins for about 3.8 months. average LDL-C levels dropped significantly from 138 to 91 mg/dL in the treated men but were virtually unchanged in control groups. Journal of Sexual Medicine first published online: 29 MAR 2014 DOI: /jsm.12521

76
Statins and Erectile Function?
Men who took statins had their erectile-function scores increased by 3.4 points, from 14.0 to 17.4 points—a 24.3% increase. The increase in erectile-function score was about one-third to one-half of that reported with phosphodiesterase inhibitors (PDE-5 inh). Pluses and Minuses: they improve endothelial function, which may improve blood flow to the penis; but on the other hand, they lower the level of cholesterol, a precursor of testosterone. Journal of Sexual Medicine first published online: 29 MAR 2014 DOI: /jsm.12521
. Pluses and Minuses: they improve endothelial function, which may improve blood flow to the penis; but on the other hand, they lower the level of cholesterol, a precursor of testosterone. Journal of Sexual Medicine first published online: 29 MAR 2014 DOI: /jsm")
77
Hepatitis C An estimated 3 million to 4 million persons in the United States are chronically infected with HCV, and approximately half are unaware of their status. These individuals may ultimately progress to advanced liver disease and/or hepatocellular cancer. However, those outcomes can be prevented by treatment, which is rapidly improving and offers the potential of a cure to more patients than has been previously possible. HCV testing is recommended at least once for persons born between 1945 and 1965.

78
Hepatitis C Risk behaviors:
Injection-drug use (current or ever, including those who injected once) Intranasal illicit drug use Risk exposures Long-term hemodialysis (ever) Getting a tattoo in an unregulated setting Healthcare, emergency medical, and public safety workers after needle sticks, sharps, or mucosal exposures to HCV-infected blood Children born to HCV-infected women Prior recipients of transfusions or organ transplants, including persons who: were notified that they received blood from a donor who later tested positive for HCV infection, received a transfusion of blood or blood components, or underwent an organ transplant before July 1992, received clotting factor concentrates produced before 1987 Were ever incarcerated
 Intranasal illicit drug use. Risk exposures. Long-term hemodialysis (ever) Getting a tattoo in an unregulated setting. Healthcare, emergency medical, and public safety workers after. needle sticks, sharps, or mucosal exposures to HCV-infected blood. Children born to HCV-infected women. Prior recipients of transfusions or organ transplants, including persons who: were notified that they received blood from a donor who later tested positive for HCV infection, received a transfusion of blood or blood components, or underwent an organ transplant before July 1992, received clotting factor concentrates produced before Were ever incarcerated.")
79
FDA Approval of Jansen’s Simeprevir - Olysio
Oct 24, 2013 The FDA Antiviral Drugs Advisory Committee voted unanimously (19 to 0) to recommend approval of the investigational protease inhibitor simeprevir (TMC435) administered once daily as a 150 mg capsule with pegylated interferon and ribavirin for the treatment of genotype 1 chronic hepatitis C in adult patients with compensated liver disease, including cirrhosis. The Advisory Committee recommended the approval of simeprevir based on analyses of data from clinical trials in patients who are treatment-naive (QUEST-1 and QUEST-2)or who have failed previous interferon-based therapy (PROMISE). FDA Approved Cost: $21,221.93/28 X 150 mg caps WAC
 to recommend approval of the investigational protease inhibitor simeprevir (TMC435) administered once daily as a 150 mg capsule with pegylated interferon and ribavirin for the treatment of genotype 1 chronic hepatitis C in adult patients with compensated liver disease, including cirrhosis. The Advisory Committee recommended the approval of simeprevir based on analyses of data from clinical trials in patients who are treatment-naive (QUEST-1 and QUEST-2)or who have failed previous interferon-based therapy (PROMISE). FDA Approved Cost: $21,221.93/28 X 150 mg caps WAC.")
80
Simeprevir - Olysio Clinical trials found simeprevir can reduce treatment time in half to 24 weeks. The medicine cured about 80 percent of patients who hadn’t been treated before compared with 50 percent of those who took pegylated interferon and ribavirin. Seventy-nine percent of simeprevir users who failed other treatments were cured compared with 37 percent who took only the older drugs,

81
Simeprevir - Olysio For patients infected with genotype 1a HCV, baseline resistance testing for the Q80K polymorphism may be considered. However, in contrast to using simeprevir to treat a genotype 1a HCV patient with PEG/RBV when the mutation markedly alters the probability of an SVR, the finding of the Q80K polymorphism does not preclude treatment with simeprevir and sofosbuvir, because the SVR rate was high in patients with genotype 1a/Q80K infection (SVR12 rate for cohort 1 was 86% [24 of 28 patients]. Thus Q80K testing can be considered but is not strongly recommended. accessed

82
Adverse Events with Simeprevir

83
Simeprevir - Olysio The most common major side effects of simeprevir were rash and photosensitivity. Panel members agreed with the FDA that the prescribing information for the drug should include a recommendation for patients to use sun protection and avoid tanning beds.

84
Simeprevir - Olysio Sulfa Allergy?
Simeprevir contains a sulfonamide moiety. In subjects with a history of sulfa allergy (n=16), no increased incidence of rash or photosensitivity reactions has been observed. However, there are insufficient data to exclude an association between sulfa allergy and the frequency or severity of adverse reactions observed with the use of Simeprevir.
, no increased incidence of rash or photosensitivity reactions has been observed. However, there are insufficient data to exclude an association between sulfa allergy and the frequency or severity of adverse reactions observed with the use of Simeprevir.")
85
FDA Approval of Gilead’s Sofosbuvir -Sovaldi
The FDA Advisory Comm. unanimously recommended approval on Oct 25,2013, the FDA approved it on Dec 6, 2013 as a “Breakthrough medication”. Cost $26,863.20/28 x 400 mg tabs WAC The FDA didn’t find any heart risks associated with sofosbuvir after Bristol-Myers and Idenix Pharmaceuticals Inc. discontinued development of drugs in the same class last year based on cardiovascular safety concerns.

86
Sofosbuvir -Sovaldi A hepatitis C virus (HCV) nucleotide analog NS5B polymerase inhibitor indicated for the treatment of chronic hepatitis C (CHC) infection as a component of a combination antiviral treatment regimen. Efficacy has been established in subjects with HCV genotype 1, 2, 3 or 4 infection, including those with hepatocellular carcinoma meeting Milan criteria (awaiting liver transplantation) and those with HCV/HIV-1 co-infection
 nucleotide analog NS5B polymerase inhibitor indicated for the treatment of chronic hepatitis C (CHC) infection as a component of a combination antiviral treatment regimen. Efficacy has been established in subjects with HCV genotype 1, 2, 3 or 4 infection, including those with hepatocellular carcinoma meeting Milan criteria (awaiting liver transplantation) and those with HCV/HIV-1 co-infection.")
87
Sofosbuvir -Sovaldi Dosage:
One 400 mg tablet taken once daily with or without food Drug Interactions: Drugs that are potent P-gp inducers in the intestine (e.g., rifampin, St. John’s wort) may significantly decrease sofosbuvir plasma concentrations and may lead to a reduced therapeutic effect
 may significantly decrease sofosbuvir plasma concentrations and may lead to a reduced therapeutic effect.")
88
Sofosbuvir - Sovaldi Safety
No serious or severe cardiac adverse events occurred in sofosbuvir-treated patients, and no treatment discontinuations occurred due to cardiac adverse events. Palpitations were the only Grade 2 event found in the SOF + RBV group. Eleven patients in the SOF + RBV group experienced Grade 1 events including palpitations, tachycardia, sinus bradycardia, extrasystoles, and ventricular extrasystoles.

89
Hepatitis C Treatment Guidelines
Summary of Recommendations for Patients Who are Initiating Therapy for HCV Infection or Who Experienced Relapse after Prior PEG/RBV Therapy, by HCV Genotype By the American Assoc for the Study of Liver Disease (AASLD)and the Infectious Diseaase Society of America (IDSA) 2014 accessed
and the Infectious Diseaase Society of America (IDSA) accessed")
90
AASLD/IDSA Summary Recommendations Hepatitis C
Genotype Recommended Alternative NOT Recommended 1 IFN eligible: SOF + PEG/RBV x 12 weeks Level 1A IFN eligible: SMV x 12 weeks + PEG/RBV x 24 For genotype 1a, baseline resistance testing for Q80K should be performed and alternative treatments considered if this mutation is present. Level IIaA TVR + PEG/RBV x 24 or 48 weeks (RGT) BOC + PEG/RBV x 28 Level IIbA IFN ineligible [1]: SOF + SMV ± RBV x 12 weeks Level 1B IFN ineligible [1]: SOF + RBV x 24 Level IIbB PEG/RBV x 48 weeks Monotherapy with PEG, RBV, or a DAA Level IIIA Do not treat decompensated cirrhosis [2] with PEG or SMV [1] [2]
 BOC + PEG/RBV x 28. Level IIbA. IFN ineligible [1]: SOF + SMV ± RBV x 12 weeks. Level 1B. IFN ineligible [1]: SOF + RBV x 24. Level IIbB. PEG/RBV x 48 weeks. Monotherapy with PEG, RBV, or a DAA Level IIIA Do not treat decompensated cirrhosis [2] with PEG or SMV. [1] [2]")
91
AASLD/IDSA Summary Recommendations Hepatitis C
“Although regimens of PEG/RBV plus telaprevir or boceprevir for 24 to 48 weeks using response guided therapy (RGT) are also FDA approved, they are markedly inferior to the preferred and alternative regimens.” “These regimens are associated with their higher rates of serious adverse events (eg, anemia and rash), longer treatment duration, high pill burden, numerous drug-drug interactions, frequency of dosing, intensity of monitoring for continuation and stopping of therapy, and the requirement to be taken with food or with high-fat meals.” accessed
 are also FDA approved, they are markedly inferior to the preferred and alternative regimens. These regimens are associated with their higher rates of serious adverse events (eg, anemia and rash), longer treatment duration, high pill burden, numerous drug-drug interactions, frequency of dosing, intensity of monitoring for continuation and stopping of therapy, and the requirement to be taken with food or with high-fat meals accessed")
92
Genotype Recommended Alternative NOT Recommended 2 SOF + RBV x 12 weeks Level IA None PEG/RBV x 24 weeks Level IIbA Monotherapy with PEG, RBV, or a DAA Level IIIA Any regimen with TVR, BOC, or SMV Level IIIA 3 SOF + RBV x 24 Level IB SOF + PEG/RBV x 12 weeks Level IIaA PEG/RBV x weeks Level IIbA

93
Genotype Recommended Alternative NOT Recommended 4 IFN eligible: SOF + PEG/RBV x 12 Weeks Level IIaB IFN ineligible [1]: SOF + RBV x 24 Weeks Level IIbB SMV x 12 weeks + PEG/RBV x 24- 48 weeks Level IIbB PEG/RBV x 48 weeks Level IIbA Monotherapy with PEG, RBV, or a DAA Level IIIA Any regimen with TVR or BOC Level IIIA 5 or 6 SOF + PEG/RBV x 12 weeks Level IIaB PEG/RBV x 48 Weeks Level IIbA IFN = interferon alfa; SOF = sofosbuvir; a nucleoside analog; PEG = peginterferon alfa; RBV = ribavirin; SMV = simeprevir; TVR = telaprevir; a direct-acting agent (DAA); BOC = boceprevir; box-summary-recommendations-patients-who-are-initiating-therapy-hcv accessed
; BOC = boceprevir; box-summary-recommendations-patients-who-are-initiating-therapy-hcv accessed")
94
Cost vs. Benefit? “A panel of experts at a recent forum in San Francisco concluded that the drug offered “low value” for treating most patients, in large part because of its high price. That judgment was based partly on an assessment of clinical and cost-effectiveness prepared by a nonprofit organization that evaluates medical treatments (the Boston-based Institute for Clinical and Economic Review). The group estimated that replacing current care of infected Californians with Sovaldi-based regimens would raise drug expenditures in the state by $18 billion or more in a single year. It said projected savings from reduced medical costs in later years would not come close to offsetting that cost.” New York Times March 15, 2014
. The group estimated that replacing current care of infected Californians with Sovaldi-based regimens would raise drug expenditures in the state by $18 billion or more in a single year. It said projected savings from reduced medical costs in later years would not come close to offsetting that cost. New York Times March 15,")
95
Cost vs. Benefit? “The drug will be sold for much less in many other countries. Gilead plans to license three to five Indian drug makers to produce Sovaldi and expects the price to be perhaps $2,000 for six months of treatment through public hospitals and community clinics in India (compared with $168,000 for six months in the United States). Gilead said similar arrangements might be made in 60 low- and middle-income countries. Prices in Europe are high, but not as high as in the United States.” New York Times March 15, 2014
. Gilead said similar arrangements might be made in 60 low- and middle-income countries. Prices in Europe are high, but not as high as in the United States. New York Times March 15,")
96
Cost vs. Benefit? “What can or should be done to reduce the cost in the United States? Some experts suggest providing the drug only to patients with advanced liver disease. Others hope that other new drugs in late stages of clinical testing will be approved, adding competition that could help restrain prices. Still others urge that state Medicaid agencies, which cover a big portion of the infected people, negotiate with Gilead for lower prices or find ways to limit the drug’s use when other good options are available. There is no easy way to contain costs as expensive new drugs come along, so all of these strategies will need to be tried.” New York Times March

97
These guidelines have been endorsed by the World Stroke Organization
Am Acad Neurology Evidence-based guideline update: Prevention of stroke in nonvalvular atrial fibrillation These guidelines have been endorsed by the World Stroke Organization For patients with NVAF, which therapies that include antithrombotic medication, as compared with no therapy or with another therapy, reduce stroke risk and severity with the least risk of hemorrhage? In patients who have NVAF but no risk factors, the absolute risk of major bleeding (3%/year) is larger than the absolute reduction in stroke from anticoagulation (1.3%/year). Neurology® 2014;82:716–724
 is larger than the absolute reduction in stroke from anticoagulation (1.3%/year). Neurology® 2014;82:716–724.")
98
Am Acad Neurology Evidence-based guideline update: Prevention of stroke in nonvalvular atrial fibrillation Selection of a Specific Oral Anticoagulant To reduce the risk of stroke or subsequent stroke in patients with NVAF judged to require oral anticoagulants, clinicians should choose one of the following options: • Warfarin, target international normalized ratio (INR) 2.0–3.0 • Dabigatran 150 mg twice daily (if creatinine clearance [CrCl] > 30 mL/min) • Rivaroxaban 15 mg/day (if CrCl 30–49 mL/min) or 20 mg/day • Apixaban 5 mg twice daily (if serum creatinine < 1.5 mg/dL) or 2.5 mg twice daily (if serum creatinine > 1.5 and < 2.5 mg/dL, and body weight < 60 kg or age at least 80 years [or both]) Level B Neurology® 2014;82:716–724
 2.0–3.0 • Dabigatran 150 mg twice daily (if creatinine clearance [CrCl] > 30 mL/min) • Rivaroxaban 15 mg/day (if CrCl 30–49 mL/min) or 20 mg/day • Apixaban 5 mg twice daily (if serum creatinine < 1.5 mg/dL) or 2.5 mg twice daily (if serum creatinine > 1.5 and < 2.5 mg/dL, and body weight < 60 kg or age at least 80 years [or both]) Level B Neurology® 2014;82:716–724.")
100
Am Acad Neurology Evidence-based guideline update: Prevention of stroke in nonvalvular atrial fibrillation GI bleeding risk Clinicians might offer apixaban to patients with NVAF and GI bleeding risk who require anticoagulant medication. Level C INR monitoring Clinicians should offer dabigatran, rivaroxaban, or apixaban to patients unwilling or unable to submit to frequent periodic testing of INR levels. Neurology® 2014;82:716–724

101
Patients unsuitable for warfarin
Am Acad Neurology Evidence-based guideline update: Prevention of stroke in nonvalvular atrial fibrillation Patients unsuitable for warfarin Clinicians should offer apixaban to patients unsuitable for being treated, or unwilling to be treated, with warfarin. Level B Where apixaban is unavailable, clinicians might offer dabigatran or rivaroxaban. Level C Where oral anticoagulants are unavailable, clinicians might offer a combination of aspirin and clopidogrel. Level C Neurology® 2014;82:716–724

102
Am Acad Neurology Evidence-based guideline update: Prevention of stroke in nonvalvular atrial fibrillation Special populations Clinicians should routinely offer oral anticoagulants to elderly patients (aged > 75 years) with NVAF if there is no history of recent unprovoked bleeding or intracranial hemorrhage. Clinicians might offer oral anticoagulation to patients with NVAF who have dementia or occasional falls. However, clinicians should counsel patients or their families that the risk–benefit ratio of oral anticoagulants is uncertain in patients with NVAF who have moderate to severe dementia or very frequent falls. Neurology® 2014;82:716–724
 with NVAF if there is no history of recent unprovoked bleeding or intracranial hemorrhage. Clinicians might offer oral anticoagulation to patients with NVAF who have dementia or occasional falls. However, clinicians should counsel patients or their families that the risk–benefit ratio of oral anticoagulants is uncertain in patients with NVAF who have moderate to severe dementia or very frequent falls. Neurology® 2014;82:716–724.")
103
2014 AHA/ACC/HRS Atrial Fibrillation Guideline
Summary of Recommendations for Prevention of Thromboembolism/Stroke in Patients With AF Antithrombotic therapy selection based on risk of thromboembolism as assessed with the CHA2DS2-VASc score With prior stroke, TIA, or CHA2DS2-VASc score ≥2, oral anticoagulants recommended. Options include: Warfarin (IA) Dabigatran, rivaroxaban, or apixaban (IB) Journal of the American College of Cardiology (2014), doi: /j.jacc
 Dabigatran, rivaroxaban, or apixaban (IB) Journal of the American College of Cardiology (2014), doi: /j.jacc")
104
2014 AHA/ACC/HRS Atrial Fibrillation Guideline
Little benefit with aspirin: This quote says a lot. "No studies, with the exception of the [Stroke Prevention in Atrial Fibrillation-1] SPAF-1 trial, show benefit for aspirin alone in preventing stroke among patients with AF.“ urged to question the common practice of using aspirin in low-risk patients. "aspirin has not been studied in a low-risk AF population" AF ablation has been moved to first-line status for both paroxysmal and persistent AF patients. This welcome change aligns these guidelines with those from Europe AF ablation should not be performed in patients who cannot be treated with anticoagulants, and AF ablation should not be done with the sole intent of avoiding anticoagulation.

105
CHADS2 -> CHA2DS2VASc CHA2DS2-VASc Risk Score CHF or LVEF < 40%
1 Hypertension Age > 75 2 Diabetes Stroke/TIA/ Thromboembolism Vascular Disease Age Female CHADS2 Risk Score CHF 1 Hypertension Age > 75 Diabetes Stroke or TIA 2 From ESC AF Guidelines

106
Adjusted stroke rate %/year
CHADS2 -> CHA2DS2VASc CHA2DS2-VASc score Patients (n = 7329) Adjusted stroke rate (%/year) 1 422 1.3 2 1230 2.2 3 1730 3.2 4 1718 4.0 5 1159 6.7 6 679 9.8 7 294 9.6 8 82 9 14 15.2 CHADS2 score Patients (n = 1733) Adjusted stroke rate %/year 120 1.9 1 463 2.8 2 523 4.0 3 337 5.9 4 220 8.5 5 65 12.5 6 18.2 From ESC AF Guidelines:
 Adjusted stroke. rate (%/year) CHADS2 score. Patients. (n = 1733) Adjusted stroke rate %/year From ESC AF Guidelines:")
107
New Indication: Apixaban - Eliquis
FDA approved for the prophylaxis of hip and knee replacement surgery based upon clinical trials in adult patients undergoing elective hip (ADVANCE-3) or knee (ADVANCE-2 and ADVANCE-1) replacement surgery. A total of 11,659 patients were randomized in 3 double-blind, multi-national studies. Included in this total were 1866 patients age 75 or older, 1161 patients with low body weight (≤60 kg), 2528 patients with Body Mass Index ≥33 kg/m2, and 625 patients with severe or moderate renal impairment.
 or knee (ADVANCE-2 and ADVANCE-1) replacement surgery. A total of 11,659 patients were randomized in 3 double-blind, multi-national studies. Included in this total were 1866 patients age 75 or older, 1161 patients with low body weight (≤60 kg), 2528 patients with Body Mass Index ≥33 kg/m2, and 625 patients with severe or moderate renal impairment.")
108
Apixaban - Eliquis Apixaban was started hours post surgery while enoxaparin was started in ADVANCE hours post surgery and continued for days; in ADVANCE hours prior to surgery and continued for days and in ADVANCE 3 – 9-15 hours prior to surgery and continued for days.

109
Apixaban - Eliquis ADVANCE 1 Trial (knee) apixaban 2.5 mg BID vs. enoxaparin 30 mg BID (FDA approved enoxaparin dosage) Combined total DVT and all cause death 8.99% vs. 8.55% RR 1.02 , p - NS ADVANCE 2 Trial (knee) apixaban 2.5 mg BID vs. enoxaparin 40 mg QD (European approved dosage) Combined total DVT and all cause death 15.06% vs % RR 0.62 , p < , NNT 11 ADVANCE 3 Trial (hip) apixaban 2.5 mg BID vs. enoxaparin 40 mg QD Combined total DVT and all cause death 1.39% vs. 3.86% RR 0.36 , p < , NNT 41
 apixaban 2.5 mg BID vs. enoxaparin 40 mg QD (European approved dosage) Combined total DVT and all cause death 15.06% vs % RR 0.62 , p < , NNT 11. ADVANCE 3 Trial (hip) apixaban 2.5 mg BID vs. enoxaparin 40 mg QD. Combined total DVT and all cause death 1.39% vs. 3.86% RR 0.36 , p < , NNT 41.")
110
Apixaban - Eliquis The recommended dose of ELIQUIS is 2.5 mg taken orally twice daily. The initial dose should be taken 12 to 24 hours after surgery. In patients undergoing hip replacement surgery, the recommended duration of treatment is 35 days. In patients undergoing knee replacement surgery, the recommended duration of treatment is 12 days.

111
Rivaroxaban - Xarelto July 5, 2011 The FDA approved rivaroxaban a factor Xa inhibitor indicated for the prophylaxis of deep vein thrombosis (DVT) which may lead to pulmonary embolism (PE) in patients undergoing knee or hip replacement. The recommended dose of is 10 mg taken orally once daily with or without food. The initial dose should be taken at least 6 to 10 hours after surgery once hemostasis has been established. For patients undergoing hip replacement surgery, treatment duration of 35 days is recommended. For patients undergoing knee replacement surgery, treatment duration of 12 days is recommended.
 which may lead to pulmonary embolism (PE) in patients undergoing knee or hip replacement. The recommended dose of is 10 mg taken orally once daily with or without food. The initial dose should be taken at least 6 to 10 hours after surgery once hemostasis has been established. For patients undergoing hip replacement surgery, treatment duration of 35 days is recommended. For patients undergoing knee replacement surgery, treatment duration of 12 days is recommended.")
112
Dosage of Enoxaparin? Most of the trials (RECORD 1,2 and 3) used the European approved dose of enoxaparin 40 mg/d. Enoxaparin was usually started the evening before surgery and continued 6 to 8 h postoperatively. RECORD 4 used the US FDA approved dose of enoxaparin: 30 mg bid dosing rather than 40 mg once daily and started 12 h postoperation.
 used the European approved dose of enoxaparin 40 mg/d. Enoxaparin was usually started the evening before surgery and continued 6 to 8 h postoperatively. RECORD 4 used the US FDA approved dose of enoxaparin: 30 mg bid dosing rather than 40 mg once daily and started 12 h postoperation.")
113
Rivaroxaban - Xarelto RECORD 1 (Hip) R=1.1% vs. E=3.9%, RRR 71%, ARR 2.8%, NNT=36 RECORD 2 (Hip) R=2.0% vs. E=8.4%, RRR 76%, ARR 6.4%, NNT=16 RECORD 3 (Knee) R=9.7% vs. E=18.8%, RRR 48%, ARR 9.1%, NNT=11 RECORD 4 (Knee) US approved dosing R=6.9% vs. E=10.1%, RRR 31%, ARR 3.19%, NNT=32
 R=9.7% vs. E=18.8%, RRR 48%, ARR 9.1%, NNT=11. RECORD 4 (Knee) US approved dosing R=6.9% vs. E=10.1%, RRR 31%, ARR 3.19%, NNT=32.")
114
Dabigatran - Pradaxa In RE-COVER, the median treatment duration during the oral only treatment period was 174 days. A total of 2539 patients (30.9% patients with symptomatic PE with or without DVT and 68.9% with symptomatic DVT only) were treated with a mean age of 54.7 years. In RE-COVER II, the median treatment duration during the oral only treatment period was 174 days. A total of 2568 patients (31.8% patients with symptomatic PE with or without DVT and 68.1% with symptomatic DVT only) were treated with a mean age of 54.9 years.
 were treated with a mean age of 54.7 years. In RE-COVER II, the median treatment duration during the oral only treatment period was 174 days. A total of 2568 patients (31.8% patients with symptomatic PE with or without DVT and 68.1% with symptomatic DVT only) were treated with a mean age of 54.9 years.")
115
Dabigatran - Pradaxa Primary Efficacy Endpoint for RE-COVER and RE-COVER II –Modified ITTa Population PRADAXA 150 mg twice daily N (%) Warfarin N (%) Hazard ratio vs. warfarin (95% CI) RE-COVER N=1274 N=1265 Primary Composite Endpointb 34 (2.7) 32 (2.5) 1.05 (0.65, 1.70) Fatal PEc 1 (0.1) 3 (0.2) Symptomatic non-fatal PEc 16 (1.3) 8 (0.6) Symptomatic recurrent DVTc 17 (1.3) 23 (1.8) RE-COVER II N=1279 N=1289 30 (2.3) 1.13 (0.69, 1.85) 9 (0.7) 15 (1.2)
 Warfarin N (%) Hazard ratio vs. warfarin (95% CI) RE-COVER. N=1274. N=1265. Primary Composite Endpointb. 34 (2.7) 32 (2.5) 1.05 (0.65, 1.70) Fatal PEc. 1 (0.1) 3 (0.2) Symptomatic non-fatal PEc. 16 (1.3) 8 (0.6) Symptomatic recurrent DVTc. 17 (1.3) 23 (1.8) RE-COVER II. N=1279. N= (2.3) 1.13 (0.69, 1.85) 9 (0.7) 15 (1.2)")
116
Dabigatran - Pradaxa In the randomized, parallel group, double-blind, pivotal trial, RE-MEDY, patients received PRADAXA 150 mg twice daily or warfarin (dosed to target INR of 2 to 3) following 3 to 12 months of treatment with anticoagulation therapy for an acute VTE. The median treatment duration during the oral only treatment period was 534 days. A total of 2856 patients were treated with a mean age of 54.6 years
 following 3 to 12 months of treatment with anticoagulation therapy for an acute VTE. The median treatment duration during the oral only treatment period was 534 days. A total of 2856 patients were treated with a mean age of 54.6 years.")
117
Hazard ratio vs. warfarin (95% CI)
Dabigatran - Pradaxa Primary Efficacy Endpoint for RE-MEDY – Modified ITTa Population PRADAXA 150 mg twice daily N=1430 N (%) Warfarin N= N (%) Hazard ratio vs. warfarin (95% CI) Primary Composite Endpointb 26 (1.8) 18 (1.3) 1.44 (0.78, 2.64) Fatal PEc 1 (0.07) Symptomatic non-fatal PEc 10 (0.7) 5 (0.4) Symptomatic recurrent DVTc 17 (1.2) 13 (0.9)
 Warfarin N=1426 N (%) Hazard ratio vs. warfarin (95% CI) Primary Composite Endpointb. 26 (1.8) 18 (1.3) 1.44 (0.78, 2.64) Fatal PEc. 1 (0.07) Symptomatic non-fatal PEc. 10 (0.7) 5 (0.4) Symptomatic recurrent DVTc. 17 (1.2) 13 (0.9)")
118
Dabigatran - Pradaxa In a randomized, parallel group, double-blind, pivotal trial, RE-SONATE, patients received PRADAXA 150 mg twice daily or placebo following 6 to 18 months of treatment with anticoagulation therapy for an acute VTE. The median treatment duration was 182 days. A total of 1343 patients were treated with a mean age of 55.8 years.

119
Dabigatran - Pradaxa Primary Efficacy Endpoint for RE-SONATE – Modified ITTa Population PRADAXA 150 mg twice daily N= N (%) Placebo N= N (%) Hazard ratio vs. placebo (95% CI) Primary Composite Endpointb 3 (0.4) 37 (5.6) 0.08 (0.02, 0.25) p-value <0.0001 Fatal PE and unexplained deathc 2 (0.3) Symptomatic non-fatal PEc 1 (0.1) 14 (2.1) Symptomatic recurrent DVTc 23 (3.5)
 Placebo N=662 N (%) Hazard ratio vs. placebo (95% CI) Primary Composite Endpointb. 3 (0.4) 37 (5.6) 0.08 (0.02, 0.25) p-value < Fatal PE and unexplained deathc. 2 (0.3) Symptomatic non-fatal PEc. 1 (0.1) 14 (2.1) Symptomatic recurrent DVTc. 23 (3.5)")
120
Fluticasone furoate /Vilanterol inhalation powder –Breo Ellipta
May 10, 2013 The Food and Drug Administration today approved Breo Ellipta (fluticasone furoate and vilanterol inhalation powder) for the long-term, once-daily, maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. It is also approved to reduce exacerbations of COPD in patients with a history of exacerbations. Developed by GlaxoSmithKline, in collaboration with Theravance.
 for the long-term, once-daily, maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. It is also approved to reduce exacerbations of COPD in patients with a history of exacerbations. Developed by GlaxoSmithKline, in collaboration with Theravance.")
121
Fluticasone furoate /Vilanterol inhalation powder –Breo Ellipta
Maintenance treatment of COPD: 1 inhalation of Breo Ellipta 100 mcg/25 mcg (fluticasone furoate /vilanterol inhalation powder) once daily. Cost $ WAC The plasma half-life of the components is ~ 24 hours and 21 hours respectively FDA Box Warning as with all other LABA containing medications Asthma Related Deaths but NOT indicated for patients with asthma
 once daily. Cost $ WAC. The plasma half-life of the components is ~ 24 hours and 21 hours respectively. FDA Box Warning as with all other LABA containing medications Asthma Related Deaths but NOT indicated for patients with asthma.")
122
Fluticasone furoate /Vilanterol inhalation powder –Breo Ellipta

123
Fluticasone furoate /Vilanterol inhalation powder –Breo Ellipta
Be careful, every time you move the cover you move to the next dose!

124
Umeclidinium and Vilanterol – Anoro Ellipta Inhaler by GSK
A combination of umeclidinium, an anticholinergic (LAMA), and vilanterol, a long-acting beta2-adrenergic agonist (LABA), indicated for the long-term, once-daily, maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD). Not indicated for the relief of acute bronchospasm or for the treatment of asthma
, and vilanterol, a long-acting beta2-adrenergic agonist (LABA), indicated for the long-term, once-daily, maintenance treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD). Not indicated for the relief of acute bronchospasm or for the treatment of asthma.")
125
Umeclidinium and Vilanterol – Anoro Ellipta Inhaler
Inhalation Powder. Inhaler containing 2 double-foil blister strips of powder formulation for oral inhalation. One strip contains umeclidinium 62.5 mcg per blister and the other contains vilanterol 25 mcg per blister. The half-life of both components is about 11 hours Dose is one inhalation once a day. FDA Box WARNING: ASTHMA-RELATED DEATH Available as both a 30 dose and 7 dose institutional inhaler

126
Umeclidinium and Vilanterol – Anoro Ellipta Inhaler
Drug Interactions: Vilanterol is a CYP 3A4 substrate so use caution when patients are taking strong 3A4 inhibitors (i.e. clarithromycin, protease inhibitors, azole antifungals) Umeclidinium is a CYP 2D6 substrate (no significant interactions seen?) MAO inhibitors and Tricyclics may increase QTc Anticholinergics are likely additive to umeclidinium, caution in males with BPH Beta blockers?
 Umeclidinium is a CYP 2D6 substrate (no significant interactions seen ) MAO inhibitors and Tricyclics may increase QTc. Anticholinergics are likely additive to umeclidinium, caution in males with BPH. Beta blockers")
127
Umeclidinium and Vilanterol – Anoro Ellipta Inhaler
Both components may increase CV risk? A dose-dependent increase in heart rate was observed (~9-20 Beats per minute increase with higher than recommended doses) Adverse Effects: include pharyngitis, sinusitis, lower respiratory tract infection, constipation, diarrhea, pain in extremity, muscle spasms, neck pain, and chest pain. Cost: $269.65/ 30 doses WAC
 Adverse Effects: include pharyngitis, sinusitis, lower respiratory tract infection, constipation, diarrhea, pain in extremity, muscle spasms, neck pain, and chest pain. Cost: $269.65/ 30 doses WAC.")
128
Umeclidinium and Vilanterol – Anoro Ellipta Inhaler
CAUTION: If you open and close the cover without inhaling the medicine, you will lose the dose. The lost dose will be held in the inhaler, but it will no longer be available to be inhaled.

129
Comparison Of Long Acting Bronchodilators
POET-COPD Trial (Prevention Of Exacerbations with Tiotropium in COPD) R, DB trial of tiotropium (18 mcg QD) vs salmeterol (50 mcg BID) in 7376 patients with moderate-to-very-severe COPD X 1 year 50% receiving inhaled steroids, using PRN beta-agonists PRN Results: Tiotropium was superior: Increased time to first exacerbation days vs 145 days (p< 0.001) Reduced number of moderate or severe exacerbations – 0.64 vs 0.72 (p=0.002) Requiring oral steroids or antibiotics Reduced number of severe exacerbations – 0.09 vs 0.13 (p< 0.001) Requiring hospitalization No difference in serious adverse events NEJM 2011;364(12):
 R, DB trial of tiotropium (18 mcg QD) vs salmeterol (50 mcg BID) in 7376 patients with moderate-to-very-severe COPD X 1 year. 50% receiving inhaled steroids, using PRN beta-agonists PRN. Results: Tiotropium was superior: Increased time to first exacerbation days vs 145 days (p< 0.001) Reduced number of moderate or severe exacerbations – 0.64 vs 0.72 (p=0.002) Requiring oral steroids or antibiotics. Reduced number of severe exacerbations – 0.09 vs 0.13 (p< 0.001) Requiring hospitalization. No difference in serious adverse events NEJM 2011;364(12):")
130
© 2014 Global Initiative for Chronic Obstructive Lung Disease
Global Strategy for Diagnosis, Management and Prevention of COPD Therapeutic Options: COPD Medications Beta2-agonists Short-acting beta2-agonists (SABA) Long-acting beta2-agonists (LABA) Anticholinergics Short-acting anticholinergics (SAMA) Long-acting anticholinergics (LAMA) tiotropium or aclindinium bromide Combination short-acting beta2-agonists + anticholinergic in one inhaler (SABA/SAMA) CombiVent Respirmat Combination long-acting beta2-agonists + anticholinergic in one inhaler (LABA/LAMA) Methylxanthines (theophylline) Inhaled corticosteroids (ICS) Combination long-acting beta2-agonists + corticosteroids in one inhaler (ICS/LABA) Systemic corticosteroids Phosphodiesterase-4 inhibitors (roflumilast) © 2014 Global Initiative for Chronic Obstructive Lung Disease
 Long-acting beta2-agonists (LABA) Anticholinergics. Short-acting anticholinergics (SAMA) Long-acting anticholinergics (LAMA) tiotropium or aclindinium bromide. Combination short-acting beta2-agonists + anticholinergic in one inhaler (SABA/SAMA) CombiVent Respirmat. Combination long-acting beta2-agonists + anticholinergic in one inhaler (LABA/LAMA) Methylxanthines (theophylline) Inhaled corticosteroids (ICS) Combination long-acting beta2-agonists + corticosteroids in one inhaler (ICS/LABA) Systemic corticosteroids. Phosphodiesterase-4 inhibitors (roflumilast) © 2014 Global Initiative for Chronic Obstructive Lung Disease.")
131
© 2013 Global Initiative for Chronic Obstructive Lung Disease
Global Strategy for Diagnosis, Management and Prevention of COPD Therapeutic Options: Bronchodilators Long-acting inhaled bronchodilators are convenient and more effective for symptom relief than short-acting bronchodilators. Long-acting inhaled bronchodilators reduce exacerbations and related hospitalizations and improve symptoms and health status. Combining bronchodilators of different pharmacological classes may improve efficacy and decrease the risk of side effects compared to increasing the dose of a single bronchodilator. © 2013 Global Initiative for Chronic Obstructive Lung Disease

132
© 2014 Global Initiative for Chronic Obstructive Lung Disease
Global Strategy for Diagnosis, Management and Prevention of COPD Therapeutic Options: Inhaled Corticosteroids Regular treatment with inhaled corticosteroids improves symptoms, lung function and quality of life and reduces frequency of exacerbations for COPD patients with an FEV1 < 60% predicted. Inhaled corticosteroid therapy is associated with an increased risk of pneumonia. Withdrawal from treatment with inhaled corticosteroids may lead to exacerbations in some patients. © 2014 Global Initiative for Chronic Obstructive Lung Disease

133
© 2014 Global Initiative for Chronic Obstructive Lung Disease
Global Strategy for Diagnosis, Management and Prevention of COPD Therapeutic Options: Combination Therapy An inhaled corticosteroid combined with a long-acting beta2-agonist is more effective than the individual components in improving lung function and health status and reducing exacerbations in moderate to very severe COPD. Combination therapy is associated with an increased risk of pneumonia. Addition of a long-acting beta2-agonist/inhaled glucorticosteroid combination to an anticholinergic (tiotropium) appears to provide additional benefits. © 2014 Global Initiative for Chronic Obstructive Lung Disease
 appears to provide additional benefits. © 2014 Global Initiative for Chronic Obstructive Lung Disease.")
134
RecommendedFirst choice
Global Strategy for Diagnosis, Management and Prevention of COPD Manage Stable COPD: Pharmacologic Therapy (Medications in each box are mentioned in alphabetical order, and therefore not necessarily in order of preference.) Patient RecommendedFirst choice Alternative choice Other Possible Treatments A SAMA prn or SABA prn LAMA LABA SABA and SAMA Theophylline B LAMA and LABA SABA and/or SAMA C ICS + LABA LAMA and LABA or LAMA and PDE4-inh. or LABA and PDE4-inh. D and/or ICS + LABA and LAMA or ICS+LABA and PDE4-inh. or LAMA and PDE4-inh. Carbocysteine
 Patient. RecommendedFirst choice. Alternative choice. Other Possible. Treatments. A. SAMA prn. or. SABA prn. LAMA. LABA. SABA and SAMA. Theophylline. B. LAMA and LABA. SABA and/or SAMA. C. ICS + LABA. LAMA and LABA or. LAMA and PDE4-inh. or. LABA and PDE4-inh. D. and/or. ICS + LABA and LAMA or. ICS+LABA and PDE4-inh. or. LAMA and PDE4-inh. Carbocysteine.")
135
Exacerbations per year
Global Strategy for Diagnosis, Management and Prevention of COPD Manage Stable COPD: Pharmacologic Therapy RECOMMENDED FIRST CHOICE C D 2 or more or > 1 leading to hospital admission 1 (not leading to hospital admission) GOLD 4 ICS + LABA or LAMA ICS + LABA and/or LAMA GOLD 3 Exacerbations per year A B GOLD 2 SAMA prn or SABA prn LABA or LAMA GOLD 1 CAT < 10 mMRC 0-1 CAT > 10 mMRC > 2 © 2014 Global Initiative for Chronic Obstructive Lung Disease
 GOLD 4. ICS + LABA. or. LAMA. ICS + LABA. and/or. LAMA. GOLD 3. Exacerbations per year. A. B. GOLD 2. SAMA prn. or. SABA prn. LABA. or. LAMA. GOLD 1. CAT < 10. mMRC 0-1. CAT > 10. mMRC > 2. © 2014 Global Initiative for Chronic Obstructive Lung Disease.")
136
Exacerbations per year
Global Strategy for Diagnosis, Management and Prevention of COPD Manage Stable COPD: Pharmacologic Therapy ALTERNATIVE CHOICE C D ICS + LABA and LAMA or ICS + LABA and PDE4-inh LAMA and LABA LAMA and PDE4-inh. LAMA and LABA or LAMA and PDE4-inh LABA and PDE4-inh 2 or more or > 1 leading to hospital admission 1 (not leading to hospital admission) GOLD 4 GOLD 3 Exacerbations per year A B GOLD 2 LAMA or LABA SABA and SAMA LAMA and LABA GOLD 1 CAT < 10 mMRC 0-1 CAT > 10 mMRC > 2 © 2014 Global Initiative for Chronic Obstructive Lung Disease
 GOLD 4. GOLD 3. Exacerbations per year. A. B. GOLD 2. LAMA. or. LABA. SABA and SAMA. LAMA and LABA. GOLD 1. CAT < 10. mMRC 0-1. CAT > 10. mMRC > 2. © 2014 Global Initiative for Chronic Obstructive Lung Disease.")
137
COPD Treatment and CV Events?
A nested case-control analysis of a retrospective cohort study in Ontario compared the risk of events between older COPD patients newly prescribed inhaled long acting beta-agonists (LABA) and long acting anticholinergics (LAMA), after matching and adjusting for prognostic factors Main Outcome and Measures: An emergency department visit or a hospitalization for a cardiovascular event. JAMA Intern Med. Published online May 20, 2013
 and long acting anticholinergics (LAMA), after matching and adjusting for prognostic factors. Main Outcome and Measures: An emergency department visit or a hospitalization for a cardiovascular event. JAMA Intern Med. Published online May 20,")
138
COPD Treatment and CV Events?
Results: Of eligible patients, (28.0%) had a hospitalization or an emergency department visit for a cardiovascular event. Patients newly prescribed a LABA and/or a LAMA were found to have a greater risk of an event compared with nonuse of those medications (respective adjusted odds ratios, 1.31 [95% CI, ; P<.001] and 1.14 [ ; P=.03]). No significant difference in events between the 2 medications (adjusted odds ratio of long-acting inhaled beta-agonists compared with anticholinergics, 1.15 [95% CI, ; P=.16]). JAMA Intern Med. Published online May 20, 2013
 had a hospitalization or an emergency department visit for a cardiovascular event. Patients newly prescribed a LABA and/or a LAMA were found to have a greater risk of an event compared with nonuse of those medications (respective adjusted odds ratios, 1.31 [95% CI, ; P<.001] and 1.14 [ ; P=.03]). No significant difference in events between the 2 medications (adjusted odds ratio of long-acting inhaled beta-agonists compared with anticholinergics, 1.15 [95% CI, ; P=.16]). JAMA Intern Med. Published online May 20,")
139
COPD Treatment and CV Events?
"These results along with the previous data support the need for close monitoring of all patients with COPD who require long-acting bronchodilators, regardless of drug class," study author Gershon and colleagues state. That conclusion, however, sits ill with Woodruff who points out in the accompanying Editorial that “without a firm recommendation as to what exactly this monitoring should consist of, monitoring remains the same as it has always been—the responsibility of the treating physician”. HeartWire Clinical Cardiology May 20, 2013

140
Vortioxetine - Brintellix by Takeda
A Selective Serotonin Reuptake Inhibitor (SSRI) antidepressant approved for treatment of major depressive disorder (MDD). a 5-hydroxytryptamine 1A (5-HT1A) agonist, 5-HT1B partial agonist, 5-HT3 antagonist, 5-HT7 antagonist, and 5-HT transporter protein inhibitor net effect of these activities is an increase in the levels of 5-HT, noradrenaline, dopamine, and acetylcholine, and histamine in the ventral hippocampus and medial prefrontal cortex
 antidepressant approved for treatment of major depressive disorder (MDD). a 5-hydroxytryptamine 1A (5-HT1A) agonist, 5-HT1B partial agonist, 5-HT3 antagonist, 5-HT7 antagonist, and 5-HT transporter protein inhibitor. net effect of these activities is an increase in the levels of 5-HT, noradrenaline, dopamine, and acetylcholine, and histamine in the ventral hippocampus and medial prefrontal cortex.")
141
Vortioxetine - Brintellix
The half-life of vortioxetine is approximately 66 hours because of the large apparent volume of distribution About 98% bound to plasma proteins Metabolized mainly by cytochrome P450 (CYP 2D6) with 6 total metabolites with little to no antidepressant activity Vortioxetine and its primary metabolite have been shown to be substrates and weak inhibitors of CYP2C19, but coadministration studies have not shown clinical significance in humans. Vortioxetine has minimal renal clearance, and negligible amounts of the drug found in urine
 with 6 total metabolites with little to no antidepressant activity. Vortioxetine and its primary metabolite have been shown to be substrates and weak inhibitors of CYP2C19, but coadministration studies have not shown clinical significance in humans. Vortioxetine has minimal renal clearance, and negligible amounts of the drug found in urine.")
142
Vortioxetine - Brintellix
The labeling for vortioxetine contains the boxed warning regarding suicidal thoughts and behaviors that is required for all antidepressants. Use of antidepressants in the treatment of children, adolescents, and young adults was associated with increases in the risk of suicidal thoughts and behaviors and a trend towards reduced risk in patients 65 years and older. All patients will need to be monitored for changes in mental function, behavior, and suicidal ideation. Use of antidepressants may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Activation of mania/hypomania may occur in patients with major affective disorder.

143
Vortioxetine - Brintellix
Table 2. Adverse Events (≥ 2%) Reported in Patients in the Placebo-Controlled Studies, Listed in Order of Magnitude of Incidence Adverse Events Vortioxetine 5 mg/day (n = 1,013) Vortioxetine 10 mg/day (n = 699) Vortioxetine 15 mg/day (n = 449) Vortioxetine 20 mg/day (n = 455) Placebo (n = 1,621) Nausea 21% 26% 32% 9% Diarrhea 7% 10% 6% Dry mouth 8% Dizziness Constipation 3% 5% Vomiting 1% Flatulence 2% Abnormal dreams < 1% Pruritus
 Reported in Patients in the Placebo-Controlled Studies, Listed in Order of Magnitude of Incidence. Adverse Events. Vortioxetine 5 mg/day (n = 1,013) Vortioxetine 10 mg/day (n = 699) Vortioxetine 15 mg/day (n = 449) Vortioxetine 20 mg/day (n = 455) Placebo (n = 1,621) Nausea. 21% 26% 32% 9% Diarrhea. 7% 10% 6% Dry mouth. 8% Dizziness. Constipation. 3% 5% Vomiting. 1% Flatulence. 2% Abnormal dreams. < 1% Pruritus.")
144
Vortioxetine - Brintellix
Table 5. ASEX Incidence of Treatment Emergent Sexual Dysfunction in Patients with No Sexual Dysfunction at Baseline Vortioxetine 5 mg/day (n = 65:67) Vortioxetine 10 mg/day (n = 94:86) Vortioxetine 15 mg/day (n = 57:67) Vortioxetine 20 mg/day (n = 67:59) Placebo (n = 135:162) Females 22% 23% 33% 34% 20% Males 16% 19% 29% 14% aSample size for each group is the number of patients (females:males) without sexual dysfunction at baseline. ASEX – Arizona Sexual Experience s Scale
 Vortioxetine 10 mg/day (n = 94:86) Vortioxetine 15 mg/day (n = 57:67) Vortioxetine 20 mg/day (n = 67:59) Placebo (n = 135:162) Females. 22% 23% 33% 34% 20% Males. 16% 19% 29% 14% aSample size for each group is the number of patients (females:males) without sexual dysfunction at baseline. ASEX – Arizona Sexual Experience s Scale.")
145
Vortioxetine - Brintellix
Drug Interactions: The risk of serotonin syndrome is increased when vortioxetine is used with other drugs that affect the serotonergic neurotransmitter system (eg, SSRIs, SNRIs, triptans, buspirone, tramadol, tryptophan) Serotonn syndrome symptoms may include changes in mental status (eg, agitation, hallucinations, delirium, coma), autonomic instability (eg, tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (eg, tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or GI symptoms (eg, nausea, vomiting, diarrhea).
 Serotonn syndrome symptoms may include changes in mental status (eg, agitation, hallucinations, delirium, coma), autonomic instability (eg, tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (eg, tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or GI symptoms (eg, nausea, vomiting, diarrhea).")
146
Vortioxetine - Brintellix
Drug Interactions: Bupropion: The active metabolites of bupropion inhibit CYP2D6, the primary metabolizing agent of vortioxetine. Multiple doses of bupropion 150 mg twice daily coadministered with vortioxetine 10 mg once daily increased the steady-state plasma exposure of vortioxetine 2-fold. Reduction in vortioxetine dose by 50% or avoidance of bupropion therapy may be necessary NSAIDs: vortioxetine and NSAID,s may both inhibit platelets and increase the risk of bleeding Warfarin: vortioxetine is a weak inhibitor of CYP2C9 in vitro, which is known to metabolize warfarin. However, multiple daily doses of vortioxetine 10 mg had no effect on the steady-state pharmacokinetics or pharmacodynamics of warfarin

147
Vortioxetine - Brintellix
Dosage: The recommended starting dose is 10 mg administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/day, as tolerated, because higher doses demonstrated better treatment effects in trials conducted in the United States. The efficacy and safety of doses above 20 mg/day have not been evaluated in controlled clinical trials. A dose decrease down to 5 mg/day may be considered for patients who do not tolerate higher doses The maximum recommended dosage for known CYP2D6 poor metabolizers is 10 mg/day Vortioxetine is available as 5, 10, 15, and 20 mg immediate-release tablets Cost: $ for 30 tabs WAC

148
Vortioxetine - Brintellix
Abrupt discontinuation of antidepressant therapy can produce discontinuation symptoms (eg, headache, muscle tension, mood swings, sudden outbursts of anger, dizziness, runny nose) during the next week. Some patients experienced these types of symptoms after abrupt discontinuation of vortioxetine 15 and 20 mg/day
 during the next week. Some patients experienced these types of symptoms after abrupt discontinuation of vortioxetine 15 and 20 mg/day.")
149
Vortioxetine - Brintellix
CONCLUSION: Vortioxetine has a mechanism of action that is different from other antidepressant medications. The results from the evaluated clinical studies are mixed. Some studies show no benefit compared with placebo, while others did show a benefit. Several of the studies have used duloxetine as an active comparator, but were not designed to compare the efficacy of vortioxetine with duloxetine. Vortioxetine appears to be well tolerated. Its place in the treatment of patients with MDD, who are treatment-naive and non–treatment naive, remains to be documented.

150
Levomilnacipran -Fetzima by Forest
A Serotonin and Norepinephrine Reuptake Inhibitor (SNRI) both levomilncaipran and milnacipran bind to both serotonin and norepinephrine transporters with high affinity, but milnacipran preferentially blocks norepinephrine reuptake compared with serotonin reuptake by an approximately 3 to 1 ratio, while levomilnacipran has a 2-fold greater potency for norepinephrine.
 both levomilncaipran and milnacipran bind to both serotonin and norepinephrine transporters with high affinity, but milnacipran preferentially blocks norepinephrine reuptake compared with serotonin reuptake by an approximately 3 to 1 ratio, while levomilnacipran has a 2-fold greater potency for norepinephrine.")
151
Levomilnacipran -Fetzima
Levomilnacipran is indicated for the treatment of major depressive disorder (MDD). Milnacipran is indicated for the management of fibromyalgia. Levomilnacipran is not approved for the management of fibromyalgia. Levomilnacipran undergoes desethylation to form desethyl levomilnacipran by CYP 3A4, approximately 58% of the dose is excreted in urine as unchanged levomilnacipran. N-desethyl levomilnacipran is the major metabolite excreted in the urine and accounted for approximately 18% of the dose. The elimination T1/2 is ~12 hours vs. 6-8 hours for milnacipran
. Milnacipran is indicated for the management of fibromyalgia. Levomilnacipran is not approved for the management of fibromyalgia. Levomilnacipran undergoes desethylation to form desethyl levomilnacipran by CYP 3A4, approximately 58% of the dose is excreted in urine as unchanged levomilnacipran. N-desethyl levomilnacipran is the major metabolite excreted in the urine and accounted for approximately 18% of the dose. The elimination T1/2 is ~12 hours vs. 6-8 hours for milnacipran.")
152
Levomilnacipran -Fetzima
Table 1. FDA-Approved Indications for SNRIs Indications Levomilnacipran Duloxetine Desvenlafaxine Milnacipran Venlafaxine Chronic musculoskeletal pain X Diabetic peripheral neuropathic pain Fibromyalgia Generalized anxiety disorder Xa MDD Panic disorder Social anxiety disorder aExtended-release formulations only.

153
Levomilnacipran -Fetzima
Table 5: Summary of Results for the Primary Efficacy Endpoint MADRS Study Number Treatment Group Mean Baseline Score (SD) LS Mean Change from Baseline (SE) Placebo Substracted Diff (95%CI) Study 1 (fixed dose) Levomilnacipran (ER 40 mg/day)* 36.0 (4.1) -14.8 (1.0) -3.2 (-5.9, -0.5) Levomilnacipran (ER 80 mg/day)* 36.1 (3.9) -15.6 (1.0) -4.0 (-6.7, -1.3) Levomilnacipran (ER 120 mg/day)* 36.0 (3.9) -16.5 (1.0) -4.9 (-7.6, -2.1) 35.6 (4.5) -11.6 (1.0) -- Study 2 (fixed- dose) 30.8 (3.4) -14.6 (0.8) -3.3 (-5.5, -1.1) Levomilnacipran(ER 80 mg/day)* 31.2 (3.5) -14.4 (0.8) -3.1 (-5.3, -1.0) 31.0 (3.8) -11.3 (0.8) Study 3 (flexible-dose) Levomilnacipran (ER mg/day)* 35.0 (3.6) -15.3 (0.8) -3.1 (-5.3, -0.9) 35.2 (3.8) -12.2 (0.8)
 LS Mean Change from Baseline (SE) Placebo. Substracted Diff. (95%CI) Study 1 (fixed dose) Levomilnacipran (ER 40 mg/day)* 36.0 (4.1) (1.0) -3.2 (-5.9, -0.5) Levomilnacipran (ER 80 mg/day)* 36.1 (3.9) (1.0) -4.0 (-6.7, -1.3) Levomilnacipran (ER 120 mg/day)* 36.0 (3.9) (1.0) -4.9 (-7.6, -2.1) 35.6 (4.5) (1.0) -- Study 2 (fixed- dose) 30.8 (3.4) (0.8) -3.3 (-5.5, -1.1) Levomilnacipran(ER 80 mg/day)* 31.2 (3.5) (0.8) -3.1 (-5.3, -1.0) 31.0 (3.8) (0.8) Study 3 (flexible-dose) Levomilnacipran (ER mg/day)* 35.0 (3.6) (0.8) -3.1 (-5.3, -0.9) 35.2 (3.8) (0.8)")
154
Levomilnacipran 40 to 120 mg/day (N = 1,583)
Adverse Reactions Occurring in > 2% of Levomilnacipran-Treated Patients and ≥ 2 Times the Rate of Placebo-Treated Patients Reported in the Placebo- Controlled Trials Adverse Reactions Placebo (N = 1,040) Levomilnacipran 40 to 120 mg/day (N = 1,583) Nausea 6% 17% Constipation 3% 9% Hyperhidrosis 2% Erectile dysfunction 1% Heart rate increased Tachycardia Ejaculation disorder < 1% 5% Palpitations Vomiting Testicular pain 4% Urinary hesitation 0% Blood pressure increased Decreased appetite Hot flush Hypertension Hypotension
 Levomilnacipran 40 to 120 mg/day (N = 1,583) Nausea. 6% 17% Constipation. 3% 9% Hyperhidrosis. 2% Erectile dysfunction. 1% Heart rate increased. Tachycardia. Ejaculation disorder. < 1% 5% Palpitations. Vomiting. Testicular pain. 4% Urinary hesitation. 0% Blood pressure increased. Decreased appetite. Hot flush. Hypertension. Hypotension.")
155
Levomilnacipran -Fetzima
Increased Heart Rate and Blood Pressure: In short-term clinical studies, levomilnacipran treatment was associated with a mean increase in heart rate of 7.4 beats per minute (bpm) compared to a mean decrease of 0.3 bpm in placebo-treated patients. Heart rate increase in levomilnacipran-treated patients receiving doses of 40 mg, 80 mg and 120 mg was 7.2, 7.2, and 9.1 bpm. In the short-term, placebo-controlled MDD studies, the mean increase from initiation of treatment in systolic BP was 3 mm Hg and diastolic BP was 3.2 mm Hg, as compared to no change in the placebo group. There were no dose-related changes in systolic and diastolic blood pressure observed. In patients exposed to one-year, open-label treatment of levomilnacipran (doses range from mg once daily), the mean change from initiation of treatment in systolic BP was 3.9 mm Hg and diastolic BP was 3.1 mm Hg.
 compared to a mean decrease of 0.3 bpm in placebo-treated patients. Heart rate increase in levomilnacipran-treated patients receiving doses of 40 mg, 80 mg and 120 mg was 7.2, 7.2, and 9.1 bpm. In the short-term, placebo-controlled MDD studies, the mean increase from initiation of treatment in systolic BP was 3 mm Hg and diastolic BP was 3.2 mm Hg, as compared to no change in the placebo group. There were no dose-related changes in systolic and diastolic blood pressure observed. In patients exposed to one-year, open-label treatment of levomilnacipran (doses range from mg once daily), the mean change from initiation of treatment in systolic BP was 3.9 mm Hg and diastolic BP was 3.1 mm Hg.")
156
Levomilnacipran -Fetzima
FDA BOX WARNING: SUICIDAL THOUGHTS AND BEHAVIORS Increased risk of suicidal thinking and behavior in children, adolescents and young adults taking antidepressants. Monitor for worsening and emergence of suicidal thoughts and behaviors. (this includes daily monitoring by families and caregivers for any changes in behaviors, emergence of agitation or irritability). Not approved for use in pediatric patients
. Not approved for use in pediatric patients.")
157
Levomilnacipran -Fetzima
Drug Interactions: Monoamine Oxidase Inhibitor (MAOI), Linezolid or Methylene Blue - At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with levomilnacipran. Conversely, at least 7 days should be allowed after stopping levomilnacipran before starting an MAOI antidepressant. With linezolid or intravenous methylene blue there is an increased risk of serotonin syndrome. Strong Inhibitors of Cytochrome P450 (CYP3A4) including (ketoconazole, clarithromycin, ritonavir) – limit the dose of levomilnacipran to no more than 80 mg/day
, Linezolid or Methylene Blue - At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with levomilnacipran. Conversely, at least 7 days should be allowed after stopping levomilnacipran before starting an MAOI antidepressant. With linezolid or intravenous methylene blue there is an increased risk of serotonin syndrome. Strong Inhibitors of Cytochrome P450 (CYP3A4) including (ketoconazole, clarithromycin, ritonavir) – limit the dose of levomilnacipran to no more than 80 mg/day.")
158
Levomilnacipran -Fetzima
Serotonin Syndrome: Serotonin syndrome has been reported with SSRIs and SNRIs, both when taken alone, but especially when co-administered with other serotonergic agents (including triptans, tricyclics, fentanyl, lithium, tramadol, tryptophan, buspirone and St. John’s Wort). symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
. symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).")
159
Levomilnacipran -Fetzima
Dosage: Recommended dose: 40 mg to 120 mg once daily with or without food. Initiate dose at 20 mg once daily for 2 days and then increase to 40 mg once daily. Based on efficacy and tolerability, increase dose in increments of 40 mg at intervals of 2 or more days. The maximum recommended dose is 120 mg once daily. Take capsules whole; do not open, chew or crush. Renal Impairment: Do not exceed 80 mg once daily for moderate impairment (creatinine clearance of ml/min). Do not exceed 40 mg once daily for severe renal impairment (creatinine clearance of ml/min). Discontinuation: Reduce dose gradually whenever possible Cost: 20, 40, 80 and 120 mg caps - $194.28/30 caps all sizes WAC
. Do not exceed 40 mg once daily for severe renal impairment (creatinine clearance of ml/min). Discontinuation: Reduce dose gradually whenever possible. Cost: 20, 40, 80 and 120 mg caps - $194.28/30 caps all sizes WAC.")
160
Levomilnacipran -Fetzima
CONCLUSION: Levomilnacipran is approved only for the treatment of MDD, while the racemic mixture, milnacipran, is approved only for the treatment of fibromyalgia. There are no head-to-head clinical trials of either of these drugs or against other SNRIs in the treatment of MDD or fibromyalgia. Both drugs are SNRIs and have similar contraindications, warnings, and precautions. Without head-to-head comparisons in studies lasting longer than 11 weeks, the value of this drug is unknown.

161
Depression: Response vs. Remission
HAM-D17 Scores Depression 15 Response: 50% reduction in baseline HAM-D score or HAM-D 15 GOAL = Remission: HAM-D Score 7 Virtually complete symptom resolution HAM-D, Hamilton Depression Score 7 The HAMD17 has 17 items and the scores go up to 48. The typical depressed patient in primary care tends to have HAMD between 15 and 19 The typical Psych outpatient tends to have HAMD between 20 and high 20s. Frank E, et al. Arch Gen Psychiatry. 1991;48:

162
Measurement-Based Care
Measurement-based care (MBC) promotes the use of rating scales or questionnaires to measure symptoms, side effects and adherence at every visit as well as guide tactics to modify dosage and treatment duration. Currently, most clinicians do not routinely use specific measures of depressive symptoms at patient visits. Rather, they tend to use global measures instead of specific symptoms measures. Results from the STAR*D study show that the use of MBC may lead to greater remission rates than those seen in efficacy studies for patients with chronic depression (Am J Psychiatry 2006;163:28-40)
 promotes the use of rating scales or questionnaires to measure symptoms, side effects and adherence at every visit as well as guide tactics to modify dosage and treatment duration. Currently, most clinicians do not routinely use specific measures of depressive symptoms at patient visits. Rather, they tend to use global measures instead of specific symptoms measures. Results from the STAR*D study show that the use of MBC may lead to greater remission rates than those seen in efficacy studies for patients with chronic depression. (Am J Psychiatry 2006;163:28-40)")
163
Choice of Antidepressant Medication & Dosing
Choice of treatment should be guided by: Patient’s prior response to treatment Tolerability Psychiatric and medical comorbidity Concurrent medications Family history of response Patient preference Cost Patient education and family involvement play critical roles in ensuring patient adherence to treatment.

164
APA Practice Guideline for the Treatment of Patients With Major Depressive Disorder 3rd Ed -2010
To reduce the risk of relapse, patients who have been treated successfully with antidepressant medications in the acute phase should continue treatment with these agents for 4–9 months [I]. In general, the dose used in the acute phase should be used in the continuation phase [II].

165
APA Practice Guideline for the Treatment of Patients With Major Depressive Disorder 3rd Ed -2010
When pharmacotherapy is being discontinued, it is best to taper the medication over the course of at least several weeks [I]. To minimize the likelihood of discontinuation symptoms, patients should be advised not to stop medications abruptly and to take medications with them when they travel or are away from home [I].

166
Conjugated Estrogens + Bazedoxifene – Duavee by Pfizer
Duavee is conjugated estrogens 0.45 mg plus bazedoxifene 20 mg/ tab for treating moderate to severe hot flashes and preventing osteoporosis. Think of it as an alternative to Prempro. Bazedoxifene is added just to inhibit estrogen's endometrial effects...as an alternative to a progestin. Bazedoxifene is a selective estrogen receptor modulator (SERM)like raloxifene (Evista). But combining bazedoxifene and estrogen doesn't work better than estrogen alone to prevent bone loss. Save Duavee for women who want to use estrogen for menopausal symptoms...but need an alternative to a progestin. Adverse Effects: >5%-≥ 5%) were muscle spasms, nausea, diarrhea, dyspepsia, abdominal pain upper, oropharyngeal pain, dizziness, and neck pain Cost: $105.84/30 tabs WAC Prescriber’s Letter March 2014
like raloxifene (Evista). But combining bazedoxifene and estrogen doesn t work better than estrogen alone to prevent bone loss. Save Duavee for women who want to use estrogen for menopausal symptoms...but need an alternative to a progestin. Adverse Effects: >5%-≥ 5%) were muscle spasms, nausea, diarrhea, dyspepsia, abdominal pain upper, oropharyngeal pain, dizziness, and neck pain. Cost: $105.84/30 tabs WAC. Prescriber’s Letter March")
167
New Pen Device for Bydureon (once weekly exenatide)
March 3, 2014: U.S. FDA Approves Bydureon® Pen (exenatide extended-release for injectable suspension) for Once-Weekly Treatment of Adults with Type 2 Diabetes. Each pen contains the recommended weekly dose of 2 mg and replaces the weekly trays which required patients to mix and draw up the dose (the trays will continue to be available as well as the pens) Now from Astra Zeneca
 for Once-Weekly Treatment of Adults with Type 2 Diabetes. Each pen contains the recommended weekly dose of 2 mg and replaces the weekly trays which required patients to mix and draw up the dose (the trays will continue to be available as well as the pens) Now from Astra Zeneca.")
168
Canagliflozin - Invokana
Canagliflozin is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The FDA has asked for five postmarketing studies for the drug including a cardiovascular outcomes trial, an enhanced pharmacovigilance program, a bone safety study and two pediatric studies Available as 100 mg and 300 mg tablets Cost: $277.41/30 tabs WAC both sizes
 inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The FDA has asked for five postmarketing studies for the drug including a cardiovascular outcomes trial, an enhanced pharmacovigilance program, a bone safety study and two pediatric studies. Available as 100 mg and 300 mg tablets. Cost: $277.41/30 tabs WAC both sizes.")
169
Canagliflozin - Invokana
Mechanism of Action Sodium-glucose co-transporter 2 (SGLT2), expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Canagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, canagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose (RTG), and thereby increases urinary glucose excretion. Maximal suppression of mean RTG over the 24‑hour period was seen with the 300 mg daily dose to approximately 70 to 90 mg/dL in patients with type 2 diabetes in Phase 1 studies.
, expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Canagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, canagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose (RTG), and thereby increases urinary glucose excretion. Maximal suppression of mean RTG over the 24‑hour period was seen with the 300 mg daily dose to approximately 70 to 90 mg/dL in patients with type 2 diabetes in Phase 1 studies.")
170
Canagliflozin - Invokana
Warnings and Precautions Hypotension - canagliflozin causes intravascular volume contraction. Symptomatic hypotension can occur after initiating csanagliflozin particularly in patients with impaired renal function (eGFR less than 60 mL/min/1.73 m2), elderly patients, patients on either diuretics or medications that interfere with the renin-angiotensin-aldosterone system (e.g., angiotensin-converting-enzyme [ACE] inhibitors, angiotensin receptor blockers [ARBs]), or patients with low systolic blood pressure. Before initiating canagliflozin in patients with one or more of these characteristics, volume status should be assessed and corrected. Monitor for signs and symptoms after initiating therapy. Keep up with fluids/hydration.
, elderly patients, patients on either diuretics or medications that interfere with the renin-angiotensin-aldosterone system (e.g., angiotensin-converting-enzyme [ACE] inhibitors, angiotensin receptor blockers [ARBs]), or patients with low systolic blood pressure. Before initiating canagliflozin in patients with one or more of these characteristics, volume status should be assessed and corrected. Monitor for signs and symptoms after initiating therapy. Keep up with fluids/hydration.")
171
Canagliflozin - Invokana
Patients with Renal Impairment Canagliflozin increases serum creatinine and decreases eGFR. Patients with hypovolemia may be more susceptible to these changes. No dose adjustment is needed in patients with mild renal impairment (eGFR of 60 mL/min/1.73 m2 or greater). The dose of canagliflozin is limited to 100 mg once daily in patients with moderate renal impairment with an eGFR of 45 to less than 60 mL/min/1.73 m2. Canagliflozin should be discontinued or not be initiated in patients with an eGFR less than 45 mL/min/1.73 m2. Assessment of renal function is recommended prior to initiation of canagliflozin therapy and periodically thereafter.
. The dose of canagliflozin is limited to 100 mg once daily in patients with moderate renal impairment with an eGFR of 45 to less than 60 mL/min/1.73 m2. Canagliflozin should be discontinued or not be initiated in patients with an eGFR less than 45 mL/min/1.73 m2. Assessment of renal function is recommended prior to initiation of canagliflozin therapy and periodically thereafter.")
172
Canagliflozin - Invokana
Genital Mycotic Infections: Canagliflozin increases the risk of genital mycotic infections. Patients with a history of genital mycotic infections and uncircumcised males were more likely to develop genital mycotic infections. (10-12% in women and 3-4% in men). Urinary Tract Infections: 4-6% of patients Increased Urination/Polyuria:
. Urinary Tract Infections: 4-6% of patients. Increased Urination/Polyuria:")
173
Canagliflozin Efficacy

174
Canagliflozin - Invokana
Recommended Dosage The recommended starting dose of canagliflozin is 100 mg once daily, taken before the first meal of the day. In patients tolerating 100 mg once daily who have an eGFR of 60 mL/min/1.73 m2 or greater and require additional glycemic control, the dose can be increased to 300 mg once daily. The dose is limited to 100 mg once daily in patients with moderate renal impairment with an eGFR of 45 to less than 60 mL/min/1.73 m2. Canagliflozin should not be initiated in patients with an eGFR less than 45 mL/min/1.73 m2.

175
Dapagliflozin-Farxiga (far-SEE-guh) by Astra Zeneca
December 13, 2013 The FDA Advisory panel voted 10 to 1 in favor of approval for AstraZeneca and Bristol-Myers Squibb's diabetes drug dapagliflozin. follows the FDA's previous rejection of dapagliflozin in January 2012, primarily due to concerns about bladder cancer and liver toxicity. all the panelists strongly advised that the sponsors move forward with a planned large postmarketing trial designed to provide enough statistical power to answer outstanding questions about cardiovascular safety as well as malignancy and liver toxicity. DECLARE-TIMI 58 trial has already begun recruiting the first of an anticipated 17,150 patients.

176
Dapagliflozin-Farxiga
It is an inhibitor of the sodium-glucose cotransporter 2 (SGLT2), expressed in the proximal renal tubules, responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Dapagliflozin 10 mg per day in patients with type 2 diabetes mellitus for 12 weeks resulted in excretion of approximately 70 grams of glucose in the urine per day.
, expressed in the proximal renal tubules, responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Dapagliflozin 10 mg per day in patients with type 2 diabetes mellitus for 12 weeks resulted in excretion of approximately 70 grams of glucose in the urine per day.")
177
Dapagliflozin-Farxiga
Pharmacokinetics: Absorption – Cmax and AUC are dose proportional and peak levels occur at 2 hours after oral dosing, oral bioavailability is ~78% (91% protein bound) Metabolism - metabolism of dapagliflozin to inactive metabolites is primarily mediated by UGT1A9; UDP-glucuronosyltransferase, an enzyme of the glucuronidation pathway that transforms small lipophilic molecules, into water-soluble, excretable metabolites Elimination – primarily renal with a mean elimination T1/2 of ~12.9 hours
 Metabolism - metabolism of dapagliflozin to inactive metabolites is primarily mediated by UGT1A9; UDP-glucuronosyltransferase, an enzyme of the glucuronidation pathway that transforms small lipophilic molecules, into water-soluble, excretable metabolites. Elimination – primarily renal with a mean elimination T1/2 of ~12.9 hours.")
178
Dapagliflozin-Farxiga
Efficacy in 24 week studies Regimen Dapagliflozin N A1C (%)* FPG (mg/dl)* Weight (Kg)* Monotherapy 10mg 70 -0.7 -24.7 Add to metformin 211 -0.5 -25.5 -2.0 Initial combo with metformin 135 -17.5 -1.4 Add to glimepiride 151 -26.5 -1.5 Add to pioglitazone 140 -0.6 -24.1 -1.8 Add to sitagliptin 223 -0.48 -27.9 -1.89 Add to insulin +/- orals 194 -25.0 -1.7 * Values are placebo/active control adjusted to reflect the effect of the addition of dapagliflozin
* FPG (mg/dl)* Weight (Kg)* Monotherapy. 10mg Add to metformin Initial combo with metformin Add to glimepiride Add to pioglitazone Add to sitagliptin Add to insulin +/- orals * Values are placebo/active control adjusted to reflect the effect. of the addition of dapagliflozin.")
179
Dapagliflozin-Farxiga
Female genital mycotic infections 6.9% and 25% in women with a history of genital mycotic infections Urinary tract infections 4.3 – 5.7% Increased urination % Male genital mycotic infections 2.8% Discomfort with urination 2.1% Mean % change in lipids: increase TC 2.5% and LDL 2.9% Hypotension: especially in the elderly, those with renal dysfunction and patientss on diuretics

180
Dapagliflozin-Farxiga
Renal-related adverse reactions, including renal failure and blood creatinine increase, were more frequent in patients treated with dapagliflozin Overall population Patients (%) with at least one event: 6.7% (n=2026 up to 104 wks) 65 years of age and older: 14.0% (n=620) eGFR ≥30 and <60 mL/min/1.73 m2: 28.3% (n=251) 65 years of age and older and eGFR ≥30 and <60 mL/min/1.73 m2: 35.1% (n=134)
 with at least one event: 6.7% (n=2026 up to 104 wks) 65 years of age and older: 14.0% (n=620) eGFR ≥30 and <60 mL/min/1.73 m2: 28.3% (n=251) 65 years of age and older and eGFR ≥30 and <60 mL/min/1.73 m2: 35.1% (n=134)")
181
Dapagliflozin-Farxiga
Across 22 clinical studies, newly diagnosed cases of bladder cancer were reported in 10/6045 patients (0.17%) treated with dapagliflozin and 1/3512 patient (0.03%) treated with placebo/comparator. After excluding patients in whom exposure to study drug was less than one year at the time of diagnosis of bladder cancer, there were 4 cases with dapagliflozin and no cases with placebo/comparator. Bladder cancer risk factors and hematuria were balanced between treatment arms at baseline. There were too few cases to determine whether the emergence of these events is related to dapagliflozin.
 treated with dapagliflozin and 1/3512 patient (0.03%) treated with placebo/comparator. After excluding patients in whom exposure to study drug was less than one year at the time of diagnosis of bladder cancer, there were 4 cases with dapagliflozin and no cases with placebo/comparator. Bladder cancer risk factors and hematuria were balanced between treatment arms at baseline. There were too few cases to determine whether the emergence of these events is related to dapagliflozin.")
182
Dapagliflozin-Farxiga
Dosage: The recommended starting dose of dapagliflozin is 5 mg once daily, taken in the morning, with or without food. In patients tolerating 5 mg once daily, the dose can be increased to 10 mg once daily Cost - $277.46/30 tabs 5 or 10 mg WAC Correct volume depletion prior to starting Monitor renal function prior to initiation of therapy and periodically thereafter Should not be initiated in patients with an eGFR less than 60 mL/min/1.73 m2 Should be discontinued when eGFR is persistently less than 60 mL/min/1.73 m

183
Albiglutide – Tanzeum by GSK
April 15, 2014 the U.S. Food and Drug Administration today approved Tanzeum (albiglutide) a glucagon-like peptide-1 (GLP-1) receptor agonist for weekly subcutaneous injection to improve glycemic control, along with diet and exercise, in adults with type 2 diabetes.
 a glucagon-like peptide-1 (GLP-1) receptor agonist for weekly subcutaneous injection to improve glycemic control, along with diet and exercise, in adults with type 2 diabetes.")
184
FDA Approved GLP-1 Agonists
Table 1. Pharmacokinetic Parameters for GLP-1 Receptor Agonists Drug Tmax Vda Half-life Albiglutide 2 to 5 days 16.4 L 6 to 8 days Exenatide 2.1 hours 28.3 L 2.4 hours Exenatide extended release 2 to 5 hours; 2 weeks; and 6 to 7 weeksb ~ 2 weeksc Liraglutide 8 to 12 hours 13 L 13 hours a Vd = volume of distribution. b Multiple peaks representing release of surface-bound exenatide, hydration, and erosion of the microspheres. c Elimination rate from controlled-release dosage form.

185
FDA Approved GLP-1 Agonists
Albiglutide Exenatide Immediate Release Exenatide Extended Release Liraglutide Route Subcutaneous Injection sites Abdomen, thigh, or upper arm Initial dosage 30 mg once weekly 5 mcg twice daily 2 mg once weekly 0.6 mg once daily Usual dosage 30 to 50 mg once weekly 5 to 10 mcg twice daily 1.2 to 1.8 mg once daily Time of day Any time, independent of meals Within 60 min before the 2 main meals of the day

186
Harmony 7 Trial Albiglutide vs Liraglutide in a randomized, phase 3, open-labeled, non-inferiority, multicenter study in 812 patients with type 2 diabetes inadequately controlled with metformin, thiazolidinediones, sulfonylureas, or any combination of these oral agents. The baseline A1c was 8.16%, age was 55.6 years, BMI was 32.8 kg/m2, and duration of diabetes was 8.4 years. Patients were randomized to 32 weeks of treatment with albiglutide once weekly or liraglutide once daily. The albiglutide group started with albiglutide 30 mg once a week for 6 weeks, then the dose was increased to 50 mg once weekly for 26 weeks. The liraglutide group started with liraglutide 0.6 mg once daily for 1 week, then 1.2 mg once daily for 1 week, and finally 1.8 mg at week 3 for the remaining 29 weeks.

187
Harmony 7 Trial Results:
Change in A1c from baseline was −0.78% with albiglutide and −0.99% with liraglutide; treatment difference was 0.21%. Weight loss was −0.64 kg with albiglutide and −2.19 kg with liraglutide. Fasting plasma glucose was −22.1 mg/dL from baseline with albiglutide and −30.4 mg/dL with liraglutide (P = 0.05).
.")
188
Harmony 7 Trial GI tract adverse reactions occurred in 35.9% of the albiglutide group and 49% of the liraglutide group. Fewer patients had nausea (9.9% vs 29.2%) and vomiting (5% vs 9.3%) with albiglutide compared with liraglutide. Hypoglycemia occurred in 16.3% of the albiglutide group and 20.8% of the liraglutide group. Injection-site reactions were more common with albiglutide than liraglutide (12.9% vs 5.4%).
 and vomiting (5% vs 9.3%) with albiglutide compared with liraglutide. Hypoglycemia occurred in 16.3% of the albiglutide group and 20.8% of the liraglutide group. Injection-site reactions were more common with albiglutide than liraglutide (12.9% vs 5.4%).")
189
Albiglutide – Tanzeum The FDA is requiring the following post-marketing studies for Tanzeum: a clinical trial to evaluate dosing, efficacy, and safety in pediatric patients; a medullary thyroid carcinoma (MTC) case registry of at least 15 years duration to identify any increase in MTC incidence related to Tanzeum; a cardiovascular outcomes trial (CVOT) to evaluate the cardiovascular risk of Tanzeum in patients with high baseline risk of cardiovascular disease.
 case registry of at least 15 years duration to identify any increase in MTC incidence related to Tanzeum; a cardiovascular outcomes trial (CVOT) to evaluate the cardiovascular risk of Tanzeum in patients with high baseline risk of cardiovascular disease.")
190
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
Reviwed by FDA Endocrinologic and Metabolic Drug Advisory Committee An ultra -rapid acting dry powder insulin for inhalation to improve glycemic control in adult and pediatric patients with diabetes mellitus Type 1 and 2 who require insulin by MannKind Corp. Technosphere Insulin Inhalation Powder (TI) is composed of recombinant human insulin and fumaryl diketopiperazine (FDKP), an inert excipient. It is administered with the breath-powered, dry powder Gen2 inhaler
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/190/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "Reviwed by FDA Endocrinologic and Metabolic Drug Advisory Committee An ultra -rapid acting dry powder insulin for inhalation to improve glycemic control in adult and pediatric patients with diabetes mellitus Type 1 and 2 who require insulin by MannKind Corp. Technosphere Insulin Inhalation Powder (TI) is composed of recombinant human insulin and fumaryl diketopiperazine (FDKP), an inert excipient. It is administered with the breath-powered, dry powder Gen2 inhaler.")
191
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
Users self-administer TI by oral inhalation using the Gen2 inhaler. The to-be-marketed cartridges contain either 0.35 mg (10 U) or 0.7 mg (20 U) of insulin. The 10 U cartridge approximates 3 units of sc injected insulin (and is labeled as “3 units”) and the 20 U cartridge approximates 6 units of sc injected insulin (and is labeled as “6 units”). The Gen2 inhaler is small, discrete, easy to use, and is discarded and replaced every 15 days.
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/191/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "Users self-administer TI by oral inhalation using the Gen2 inhaler. The to-be-marketed cartridges contain either 0.35 mg (10 U) or 0.7 mg (20 U) of insulin. The 10 U cartridge approximates 3 units of sc injected insulin (and is labeled as 3 units ) and the 20 U cartridge approximates 6 units of sc injected insulin (and is labeled as 6 units ). The Gen2 inhaler is small, discrete, easy to use, and is discarded and replaced every 15 days.")
192
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
No cleaning is required. As a breath-powered inhaler, it relies on a person’s inhalation effort to reproducibly deliver TI to the pulmonary tract. Replaced every 15 days.
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/192/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "No cleaning is required. As a breath-powered inhaler, it relies on a person’s inhalation effort to reproducibly deliver TI to the pulmonary tract. Replaced every 15 days.")
193
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
Technosphere insulin inhaltion has a more rapid onset of insulin action and shorter duration of effect, as measured by the glucose infusion rate (GIR) during a euglycemic clamp study , when compared with sc Human Regular or insulin lispro. Some data suggest that there is less weight gain (even a modest loss in some patients) and less risk of hypoglycemia (1.7% vs. 2.3%) than with injectable insulin but there was also more cough (26.9% vs. 5.2%), a slightly greater reduction in FEV1 (mean difference, was -40 mL at 3 months and -45 mL at 24 months), and disappeared upon discontinuation of TI therapy with the inhaled insulin. Patients with asthma and COPD should not be given inhaled insulin, proposed labeling will say contraindicated!
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/193/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "Technosphere insulin inhaltion has a more rapid onset of insulin action and shorter duration of effect, as measured by the glucose infusion rate (GIR) during a euglycemic clamp study , when compared with sc Human Regular or insulin lispro. Some data suggest that there is less weight gain (even a modest loss in some patients) and less risk of hypoglycemia (1.7% vs. 2.3%) than with injectable insulin but there was also more cough (26.9% vs. 5.2%), a slightly greater reduction in FEV1 (mean difference, was -40 mL at 3 months and -45 mL at 24 months), and disappeared upon discontinuation of TI therapy with the inhaled insulin. Patients with asthma and COPD should not be given inhaled insulin, proposed labeling will say contraindicated!")
194
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/194/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "AFREZZA® (insulin human [rDNA origin]) Inhalation Powder")
195
AFREZZA® (insulin human [rDNA origin]) Inhalation Powder
On April 1, 2014 the FDA Endocrinologic and Metabolic Drug Advisory Committee voted 13-1 with one abstention to approve Afrezza for Type 1 diabetics, and 14-0 with one abstention for Type 2 diabetics. While the FDA isn’t required to follow the panel’s recommendation, it rarely contradicts it. An official determination is expected by April 15. Prior to the meeting FDA staff report worried investors in the stock, as it noted the drug’s potential risk of causing lung infections and bronchial spasms and coughing that caused some patients to stop treatment. Staff also questioned the accuracy of insulin doses in MannKind’s clinical studies. Concern was also raised about the potential for lung cancer with long-term therapy as was seen with Pfizer’s Exubera.
![AFREZZA® (insulin human [rDNA origin]) Inhalation Powder](http://slideplayer.com/slide/1595190/5/images/195/AFREZZA%C2%AE+%28insulin+human+%5BrDNA+origin%5D%29+Inhalation+Powder.jpg "On April 1, 2014 the FDA Endocrinologic and Metabolic Drug Advisory Committee voted 13-1 with one abstention to approve Afrezza for Type 1 diabetics, and 14-0 with one abstention for Type 2 diabetics. While the FDA isn’t required to follow the panel’s recommendation, it rarely contradicts it. An official determination is expected by April 15. Prior to the meeting FDA staff report worried investors in the stock, as it noted the drug’s potential risk of causing lung infections and bronchial spasms and coughing that caused some patients to stop treatment. Staff also questioned the accuracy of insulin doses in MannKind’s clinical studies. Concern was also raised about the potential for lung cancer with long-term therapy as was seen with Pfizer’s Exubera.")
196
ORALAIR® by Stallergenes (Sweet Vernal, Orchard, Perennial Rye, Timothy, and Kentucky Blue Grass Mixed Pollens Allergen Extract) ORALAIR is a sublingual tablet of 5 grass pollen allergen extracts indicated as immunotherapy for the treatment of grass pollen-induced allergic rhinitis with or without conjunctivitis confirmed by positive skin test or in vitro testing for pollen-specific IgE antibodies for any of the five grass species contained in this product. ORALAIR is approved for use in persons 10 through 65 years of age.

197
Oralair Box Warning: Severe Allergic Reactions
ORALAIR can cause life-threatening allergic reactions such as anaphylaxis and severe laryngopharyngeal restriction. Do not administer ORALAIR to patients with severe, unstable or uncontrolled asthma. Observe patients in the office for at least 30 minutes following the initial dose. Prescribe auto-injectable epinephrine, instruct and train patients on its appropriate use, and instruct patients to seek immediate medical care upon its use. ORALAIR may not be suitable for patients with certain underlying medical conditions that may reduce their ability to survive a serious allergic reaction. ORALAIR may not be suitable for patients who may be unresponsive to epinephrine or inhaled bronchodilators, such as those taking beta-blokers

198
Oralair sublingual tablet Dosing
For adults 18 through 65 years of age, the dose is 300 IR (index of reactivity) daily. For children and adolescents 10 through 17 years of age, the dose is increased over the first three days from 100 IR on day one, 2 x 100 IR on day two and then 300 IR on day three and after. Comes as 100 and 300 IR sublingual tablets Remove the ORALAIR tablet from the blister just prior to dosing. Place the ORALAIR tablet immediately under the tongue until complete dissolution for at least 1 minute before swallowing. Wash hands after handling the ORALAIR tablet. Do not take the ORALAIR tablet with food or beverage. To avoid swallowing allergen extract, food or beverage should not be taken for 5 minutes following dissolution of the tablet. Initiate treatment 4 months before the expected onset of each grass pollen season and maintain it throughout the grass pollen season.
 daily. For children and adolescents 10 through 17 years of age, the dose is increased over the first three days from 100 IR on day one, 2 x 100 IR on day two and then 300 IR on day three and after. Comes as 100 and 300 IR sublingual tablets. Remove the ORALAIR tablet from the blister just prior to dosing. Place the ORALAIR tablet immediately under the tongue until complete dissolution for at least 1 minute before swallowing. Wash hands after handling the ORALAIR tablet. Do not take the ORALAIR tablet with food or beverage. To avoid swallowing allergen extract, food or beverage should not be taken for 5 minutes following dissolution of the tablet. Initiate treatment 4 months before the expected onset of each grass pollen season and maintain it throughout the grass pollen season.")
199
Oralair sublingual tablet
Contraindicated in patients with: Severe, unstable or uncontrolled asthma History of any severe systemic allergic reaction History of any severe local reaction to sublingual allergen immunotherapy Hypersensitivity to any of the inactive ingredients (mannitol, microcrystalline cellulose, croscarmellose sodium, colloidal anhydrous silica, magnesium stearate and lactose monohydrate)
")
200
Oralair sublingual tablet
Adverse Effect IR (n=1038) Placebo (n=840) Ear pruritus % 0.6% Throat irritation % 3.7% Cough % 5.9% Oropharyngeal pain % 3.7% Pharyngeal edema % 0.1% Oral pruritus % 5.0% Edema mouth % 0.6% Tongue pruritus 7.9% 0.7% Lip edema % 0.4% Paraesthesia oral 4.3% 1.0% Tongue edema 2.7% 0.1% Urticaria % 1.5%
 Placebo (n=840) Ear pruritus 8.4% 0.6% Throat irritation 22.0% 3.7% Cough 7.3% 5.9% Oropharyngeal pain 5.1% 3.7% Pharyngeal edema 3.8% 0.1% Oral pruritus 25.1% 5.0% Edema mouth 8.2% 0.6% Tongue pruritus 7.9% 0.7% Lip edema 4.4% 0.4% Paraesthesia oral 4.3% 1.0% Tongue edema 2.7% 0.1% Urticaria 2.3% 1.5%")
201
Oralair sublingual tablet
Clinical Trials: The efficacy of ORALAIR for the treatment of grass pollen-induced allergic rhinoconjunctivitis was investigated in five double-blind, placebo-controlled clinical trials: four natural field studies and an environmental exposure chamber study. The daily Combined Score (CS, range: 0-3) equally weights symptoms and rescue medication use. The daily Rhinoconjunctivitis Total Symptom Score (RTSS, range 0-18) is the total of the six individual symptom scores (sneezing, rhinorrhea, nasal pruritus, nasal congestion, ocular pruritus and watery eyes) each graded by participants on a 0 (no symptoms) to 3 (severe symptoms) scale. The daily Rescue Medication Score (RMS, range 0-3) grades the intake of rescue medication as 0 = absent, 1 = antihistamine, 2 = nasal corticosteroid, 3 = oral corticosteroid
 equally weights symptoms and rescue medication use. The daily Rhinoconjunctivitis Total Symptom Score (RTSS, range 0-18) is the total of the six individual symptom scores (sneezing, rhinorrhea, nasal pruritus, nasal congestion, ocular pruritus and watery eyes) each graded by participants on a 0 (no symptoms) to 3 (severe symptoms) scale. The daily Rescue Medication Score (RMS, range 0-3) grades the intake of rescue medication as 0 = absent, 1 = antihistamine, 2 = nasal corticosteroid, 3 = oral corticosteroid.")
202
Oralair Efficacy Data In the US study, 473 adults aged 18 through 65 years received ORALAIR or placebo, starting approximately four months prior to the expected onset of the grass-pollen season and continuing for the duration of the pollen season. The results of the analysis of the daily Combined Score (CS), daily Rhinoconjunctivitis Total Symptom Score (RTSS), and daily Rescue Medication Score (RMS)
, daily Rhinoconjunctivitis Total Symptom Score (RTSS), and daily Rescue Medication Score (RMS)")
203
Oralair US Efficacy Trial
Daily CS (primary outcome) 0.32 Rx vs Placebo (-0.13 / -28.2% diff) 95%CI [-43.4%;-13.0%] Daily RTSS 3.21 Rx vs Placebo (-0.95 / -22.9% diff) 95%% CI [-38.2%;-7.5%] Daily RMS 0.11 Rx vs Placebo (-0.09 / -46.5% diff) 95% CI [-73.9%;-19.2%]
 0.32 Rx vs Placebo (-0.13 / -28.2% diff) 95%CI [-43.4%;-13.0%] Daily RTSS Rx vs Placebo (-0.95 / -22.9% diff) 95%% CI [-38.2%;-7.5%] Daily RMS Rx vs Placebo (-0.09 / -46.5% diff) 95% CI [-73.9%;-19.2%]")
204
Oralair Long-term Efficacy Trial
In this study, adults received ORALAIR or placebo according to two different treatment regimens. A total of 426 subjects received ORALAIR or placebo starting approximately 4 months prior to the grass pollen season and continuing for the entire season. Subjects were treated for three consecutive grass pollen seasons (Year 1 to Year 3). Primary outcome was daily combined score (CS)
. Primary outcome was daily combined score (CS)")
205
Oralair Long-term Efficacy Trial
Year 1 ( n= 188 Rx and 205 placebo) CS 0.56 Rx vs Placebo (-0.11 /-16.4% diff) 95% CI [-27.0%;-5.8%] Year 2 (n= 160 Rx and 172 placebo) CS 0.35 Rx vs Placebo (-0.21/-38.0% diff) 95% CI [-53.4%;-22.6%] Year 3 (n= 149 Rx and 165 placebo) CS 0.31 Rx vs Placebo (-0.19/-38.3% diff) 95% CI [-54.7%;-22.0%]
 CS 0.56 Rx vs Placebo (-0.11 /-16.4% diff) 95% CI [-27.0%;-5.8%] Year 2 (n= 160 Rx and 172 placebo) CS 0.35 Rx vs Placebo (-0.21/-38.0% diff) 95% CI [-53.4%;-22.6%] Year 3 (n= 149 Rx and 165 placebo) CS 0.31 Rx vs Placebo (-0.19/-38.3% diff) 95% CI [-54.7%;-22.0%]")
206
Oralair Pediatric Efficacy Trial
278 children and adolescents received ORALAIR (n=131) or placebo (n=135) starting approximately 4 months prior to the grass-pollen season and continuing for the duration of the pollen season.
 or placebo (n=135) starting approximately 4 months prior to the grass-pollen season and continuing for the duration of the pollen season.")
207
Oralair Pediatric Efficacy Trial
Daily CS 0.44 Rx vs placebo (-0.19/-30.1% diff) 95% CI [-46.9%;-13.2%] Daily RTSS 2.52 Rx vs placebo (-1.11/-30.6% diff) 95% CI [-47.0%;-14.1%] Daily RMS 0.46 Rx vs placebo (-0.19/-29.5% diff) 95% CI [-50.9%;-8.0%]
 95% CI [-46.9%;-13.2%] Daily RTSS Rx vs placebo (-1.11/-30.6% diff) 95% CI [-47.0%;-14.1%] Daily RMS Rx vs placebo (-0.19/-29.5% diff) 95% CI [-50.9%;-8.0%]")
208
Oralair Allergen Environmental Chamber Study
89 adults with grass pollen-associated allergic rhinoconjunctivitis were challenged with four of the five grass pollens contained in ORALAIR at baseline and after 4 months of treatment with ORALAIR (n=45) or placebo (n=44) The average Rhinoconjunctivitis Total Symptom Score (RTSS) of each group during the 4 hours of the allergen challenge was assessed; use of rescue medication was not permitted.
 or placebo (n=44) The average Rhinoconjunctivitis Total Symptom Score (RTSS) of each group during the 4 hours of the allergen challenge was assessed; use of rescue medication was not permitted.")
209
Oralair Allergen Environmental Chamber Study
Average RTSS 4.88 Rx vs placebo (-1.97/-28.7% diff) 955 CI [-43.7%;-13.7%]
 955 CI [-43.7%;-13.7%]")
210
Timothy Grass Pollen Allergen Extract- Grastek by Merck
April 15, 2014 the FDA approved the new sublingual timothy grass pollen extract following a unanimous FDA Allergenic Products Advisory Committee vote to recommend Grastek for approval on December 12, 2013. Sublingual immunotherapy (SLIT) indicated for the treatment of Timothy grass pollen-induced allergic rhinitis, with or without conjunctivitis, in individuals aged 5 to 65 years confirmed by positive skin test or in vitro testing for pollen-specific IgE antibodies for Timothy grass or cross-reactive grass pollens. Grastek has been on the market in Europe since 2006 under the trade name Grazax.
 indicated for the treatment of Timothy grass pollen-induced allergic rhinitis, with or without conjunctivitis, in individuals aged 5 to 65 years. confirmed by positive skin test or in vitro testing for pollen-specific IgE antibodies for Timothy grass or cross-reactive grass pollens. Grastek has been on the market in Europe since 2006 under the trade name Grazax.")
211
Timothy Grass Pollen Allergen Extract- Grastek
WARNING: SEVERE ALLERGIC REACTIONS GRASTEK can cause life-threatening allergic reactions such as anaphylaxis and severe laryngopharyngeal restriction. Do not administer GRASTEK to patients with severe, unstable or uncontrolled asthma. Observe patients in the office for at least 30 minutes following the initial dose. Prescribe auto-injectable epinephrine, instruct and train patients on its appropriate use, and instruct patients to seek immediate medical care upon its use. GRASTEK may not be suitable for patients with certain underlying medical conditions that may reduce their ability to survive a serious allergic reaction. (5.2) GRASTEK may not be suitable for patients who may be unresponsive to epinephrine or inhaled bronchodilators, such as those taking beta-blockers.
 GRASTEK may not be suitable for patients who may be unresponsive to epinephrine or inhaled bronchodilators, such as those taking beta-blockers.")
212
Timothy Grass Pollen Allergen Extract- Grastek
CONTRAINDICATIONS: Severe, unstable or uncontrolled asthma. History of any severe systemic allergic reaction or any severe local reaction to sublingual allergen immunotherapy. A history of eosinophilic esophagitis. Hypersensitivity to any of the inactive ingredients contained in this product.

213
Timothy Grass Pollen Allergen Extract- Grastek
Total Combined Scores (TCS), Rhinoconjunctivitis Daily Symptom Scores (DSS), and Daily Medication Scores (DMS) During the Grass Pollen Season Endpoint* GRASTEK Placebo Treatment Difference Difference Relative to Placebo (N)† ‡Score (N)† ‡Score (GRASTEK – Placebo) Estimate (95% CI) TCS Entire Season (629) (672) % (-36.0, -13.0) TCS Peak Season (620) (663) % (-39.0, -15.0) DSS Entire Season (629) (672) % (-32.0, -10.0) DMS Entire Season(629) (672) % (-49.3, -20.8) Efficacy was established by self-reporting of rhinoconjunctivitis daily symptom scores (DSS) and daily medication scores (DMS). Daily rhinoconjunctivitis symptoms included four nasal symptoms (runny nose, stuffy nose, sneezing, and itchy nose), and two ocular symptoms (gritty/itchy eyes and watery eyes). The rhinoconjunctivitis symptoms were measured on a scale of 0 (none) to 3 (severe). Subjects in clinical trials were allowed to take symptom-relieving medications (including systemic and topical antihistamines and topical and oral corticosteroids) as needed.
, Rhinoconjunctivitis Daily Symptom Scores (DSS), and Daily Medication Scores (DMS) During the Grass Pollen Season. Endpoint* GRASTEK Placebo Treatment Difference Difference Relative to Placebo. (N)† ‡Score (N)† ‡Score (GRASTEK – Placebo) Estimate (95% CI) TCS Entire Season (629) 3.24 (672) % (-36.0, -13.0) TCS Peak Season (620) 3.33 (663) % (-39.0, -15.0) DSS Entire Season (629) 2.49 (672) % (-32.0, -10.0) DMS Entire Season(629) 0.88 (672) % (-49.3, -20.8) Efficacy was established by self-reporting of rhinoconjunctivitis daily symptom scores (DSS) and daily medication scores (DMS). Daily rhinoconjunctivitis symptoms included four nasal symptoms (runny nose, stuffy nose, sneezing, and itchy nose), and two ocular symptoms (gritty/itchy eyes and watery eyes). The rhinoconjunctivitis symptoms were measured on a scale of 0 (none) to 3 (severe). Subjects in clinical trials were allowed to take symptom-relieving medications (including systemic and topical antihistamines and topical and oral corticosteroids) as needed.")
214
Timothy Grass Pollen Allergen Extract- Grastek
Adverse Reactions Reported in ≥1% of Adults Treated with GRASTEK Adverse Reaction GRASTEK (N=1669) PLACEBO (N=1645) Nervous System Disorders Headache % 1.3% Ear and Labyrinth Disorders Ear pruritus 12.5% 1.1% Respiratory, Thoracic and Mediastinal Disorders Throat irritation % 2.8% Pharyngeal edema 3.4% 0.1% Dry throat % 0.4% Oropharyngeal pain 1.6% 1.0% Nasal discomfort % 1.0% Throat tightness % 0.2% Dyspnea % 0.4% Gastrointestinal Disorders Oral pruritus % 3.5% Mouth edema % 0.8% Paraesthesia oral % 2.0% Tongue pruritus % 0.5%
 PLACEBO (N=1645) Nervous System Disorders. Headache 2.1% 1.3% Ear and Labyrinth Disorders Ear pruritus 12.5% 1.1% Respiratory, Thoracic and Mediastinal Disorders. Throat irritation 22.6% 2.8% Pharyngeal edema 3.4% 0.1% Dry throat 1.7% 0.4% Oropharyngeal pain 1.6% 1.0% Nasal discomfort 1.6% 1.0% Throat tightness 1.4% 0.2% Dyspnea 1.1% 0.4% Gastrointestinal Disorders. Oral pruritus 26.7% 3.5% Mouth edema 11.1% 0.8% Paraesthesia oral 9.8% 2.0% Tongue pruritus 5.7% 0.5%")
215
Timothy Grass Pollen Allergen Extract- Grastek
Pediatric safety data are based on 3 clinical trials which randomized 881 subjects between 5 and 17 years of age with grass pollen induced rhinitis with or without conjunctivitis. Overall, 445 subjects received at least one dose of GRASTEK’ The most common adverse reactions in pediatric subjects treated with GRASTEK were oral pruritus (24.4% vs 2.1% placebo), throat irritation (21.3% vs 2.5%) and mouth edema (9.8% vs 0.2%). The percentage of subjects who discontinued from the clinical trials because of an adverse reaction while exposed to GRASTEK or placebo was 6.3% and 0.7%, respectively.
, throat irritation (21.3% vs 2.5%) and mouth edema (9.8% vs 0.2%). The percentage of subjects who discontinued from the clinical trials because of an adverse reaction while exposed to GRASTEK or placebo was 6.3% and 0.7%, respectively.")
216
Timothy Grass Pollen Allergen Extract- Grastek
In European post-approval studies which included 1,666 patients treated with GRASTEK (marketed under the name GRAZAX), reported serious adverse reactions assessed as related to GRASTEK use included anaphylactic reaction, asthma exacerbation, hoarseness, laryngitis, oral ulceration, and ulcerative colitis exacerbation.
, reported serious adverse reactions assessed as related to GRASTEK use included anaphylactic reaction, asthma exacerbation, hoarseness, laryngitis, oral ulceration, and ulcerative colitis exacerbation.")
217
Timothy Grass Pollen Allergen Extract- Grastek
Initiate treatment at least 12 weeks before the expected onset of each grass pollen season and continue treatment throughout the season. For sustained effectiveness for one grass pollen season after cessation of treatment, GRASTEK may be taken daily for three consecutive years (including the intervals between the grass pollen seasons). GRASTEK is available as 2800 Bioequivalent Allergy Unit (BAU) tablets for sublingual administration only. Prescribe auto-injectable epinephrine to patients prescribed GRASTEK and instruct them in the proper use of emergency self-injection of epinephrine
. GRASTEK is available as 2800 Bioequivalent Allergy Unit (BAU) tablets for sublingual administration only. Prescribe auto-injectable epinephrine to patients prescribed GRASTEK and instruct them in the proper use of emergency self-injection of epinephrine.")
218
Timothy Grass Pollen Allergen Extract- Grastek
Administer the first dose of GRASTEK in a healthcare setting under the supervision of a physician with experience in the diagnosis and treatment of allergic diseases. After receiving the first dose of GRASTEK, observe the patient for at least 30 minutes to monitor for signs or symptoms of a severe systemic or a severe local allergic reaction. If the patient tolerates the first dose, the patient may take subsequent doses at home. Take the tablet from the blister unit after carefully removing the foil with dry hands. Place the tablet immediately under the tongue. Allow it to remain there until completely dissolved. Do not swallow for at least 1 minute. Wash hands after handling the tablet. Do not take the tablet with food or beverage. Food or beverage should not be taken for the following 5 minutes after taking the tablet.

219
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek by Merck
April 17, 2014 FDA approved Ragwitek, the first allergen extract administered under the tongue (sublingually) to treat short ragweed pollen induced allergic rhinitis (hay fever), with or without conjunctivitis (eye inflammation), in adults 18 years through 65 years of age. Treatment with Ragwitek is started 12 weeks before the start of ragweed pollen season and continued throughout the season. The first dose is taken in a health care professional’s office where the patient is to be observed for at least 30 minutes for potential adverse reactions. After the first dose, patients can take Ragwitek at home.
 to treat short ragweed pollen induced allergic rhinitis (hay fever), with or without conjunctivitis (eye inflammation), in adults 18 years through 65 years of age. Treatment with Ragwitek is started 12 weeks before the start of ragweed pollen season and continued throughout the season. The first dose is taken in a health care professional’s office where the patient is to be observed for at least 30 minutes for potential adverse reactions. After the first dose, patients can take Ragwitek at home.")
220
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek
The safety and effectiveness of Ragwitek was evaluated in studies conducted in the United States and internationally. Safety was assessed in approximately 1,700 adults. The most commonly reported adverse reactions by patients treated with Ragwitek were itching in the mouth and ears and throat irritation. Of the 1,700 adults, about 760 were evaluated to determine effectiveness. Some patients received Ragwitek; others received an inactive substitute (placebo). The patients reported their symptoms and additional medications needed to get through the allergy season. During treatment for one ragweed pollen season, patients who received Ragwitek experienced approximately a 26 percent reduction in symptoms and the need for medications compared to those who received a placebo.
. The patients reported their symptoms and additional medications needed to get through the allergy season. During treatment for one ragweed pollen season, patients who received Ragwitek experienced approximately a 26 percent reduction in symptoms and the need for medications compared to those who received a placebo.")
221
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek
The dosage of the tablets proposed for use in the US is 12 Amb a 1 – U of extract derived from short ragweed (Ambrosia artemisiifolia) pollen. (Equivalent to about 1 mcg of Amb a 1) To measure symptoms, the sponsors used the average rhinoconjunctivitis Daily Symptom Score (DSS). The DSS is the sum of six individual rhinoconjunctivitis symptom scores with possible values of 0 (absent) to 3 (severe). The six symptoms that are scored are: runny nose, blocked nose, sneezing, itchy nose, gritty feeling/red/itchy eyes, and watery eyes. The maximum DSS is 18.
 pollen. (Equivalent to about 1 mcg of Amb a 1) To measure symptoms, the sponsors used the average rhinoconjunctivitis Daily Symptom Score (DSS). The DSS is the sum of six individual rhinoconjunctivitis symptom scores with possible values of 0 (absent) to 3 (severe). The six symptoms that are scored are: runny nose, blocked nose, sneezing, itchy nose, gritty feeling/red/itchy eyes, and watery eyes. The maximum DSS is 18.")
222
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek
To measure medication use, the sponsors use the average rhinoconjunctivitis Daily Medication Score (DMS). The DMS is the sum of scores that are assigned to each medication (IE antihistamines 6, topical steroids 8 and systemic steroids 16 with a max scores of 36) The primary efficacy endpoint for each of the studies was the total combined score (TCS) over the peak of ragweed season. The TCS is the sum of DSS (maximum 18) and DMS (maximum 36). The maximum TCS is 54.
. The DMS is the sum of scores that are assigned to each medication (IE antihistamines 6, topical steroids 8 and systemic steroids 16 with a max scores of 36) The primary efficacy endpoint for each of the studies was the total combined score (TCS) over the peak of ragweed season. The TCS is the sum of DSS (maximum 18) and DMS (maximum 36). The maximum TCS is 54.")
223
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek
FDA’s assessment of meaningful differences for allergen immunotherapy: a percent difference in average scores ≤ - 15%, and a 95% CIUL ≤ -10% for the TCS during the peak of ragweed season and over the entire ragweed season.

224
Extract from Short Ragweed (Ambrosia artemisiifolia) Pollen – Ragwitek
Treatment –related adverse events were reported at a higher frequency among the 1707 subjects treated over 28 days with RAGWITEK 12 Amb a 1-U compared to the 757 placebo subjects (56.5% RAGWITEK, 37.6% placebo). The most commonly reported treatment -related adverse events were: Oral pruritus (11.0% RAGWITEK; 2.0 % placebo), Ear pruritus (10.7% RAGWITEK; 1.1% placebo), Throat irritation (17.0% RAGWITEK; 3.8% placebo), and Mouth edema (6.1% RAGWITEK; 0.5% placebo).
. The most commonly reported treatment -related adverse events were: Oral pruritus (11.0% RAGWITEK; 2.0 % placebo), Ear pruritus (10.7% RAGWITEK; 1.1% placebo), Throat irritation (17.0% RAGWITEK; 3.8% placebo), and. Mouth edema (6.1% RAGWITEK; 0.5% placebo).")
Similar presentations

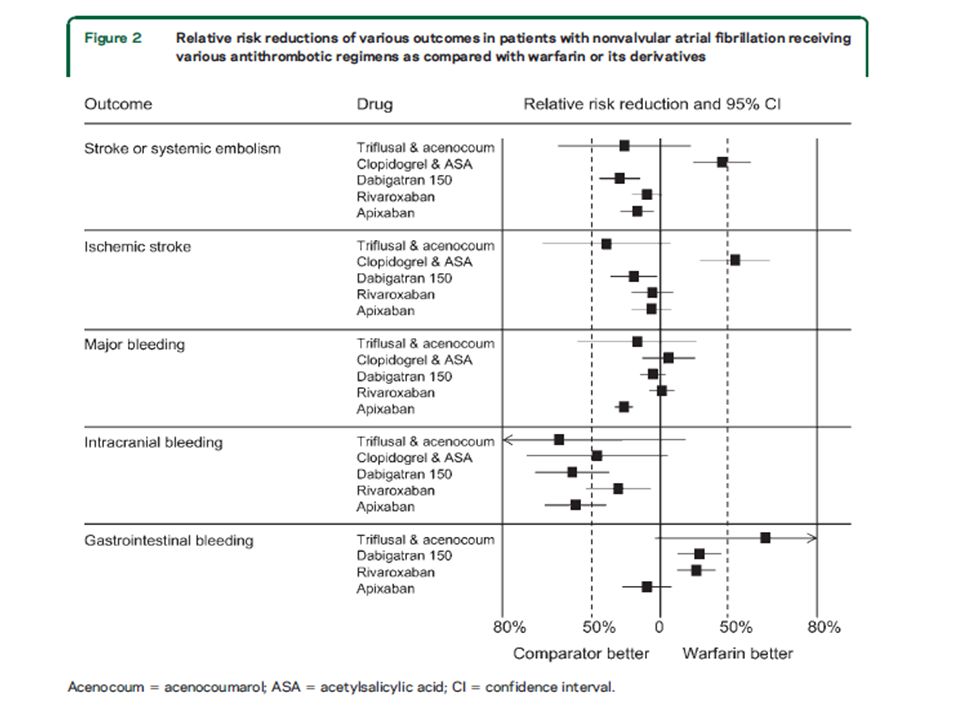







 patients Policy.>")







 Motion Controller Design for A Class of Second-order Systems Center for Self-Organizing Intelligent.>")



